Dr.Afraa Mamoori
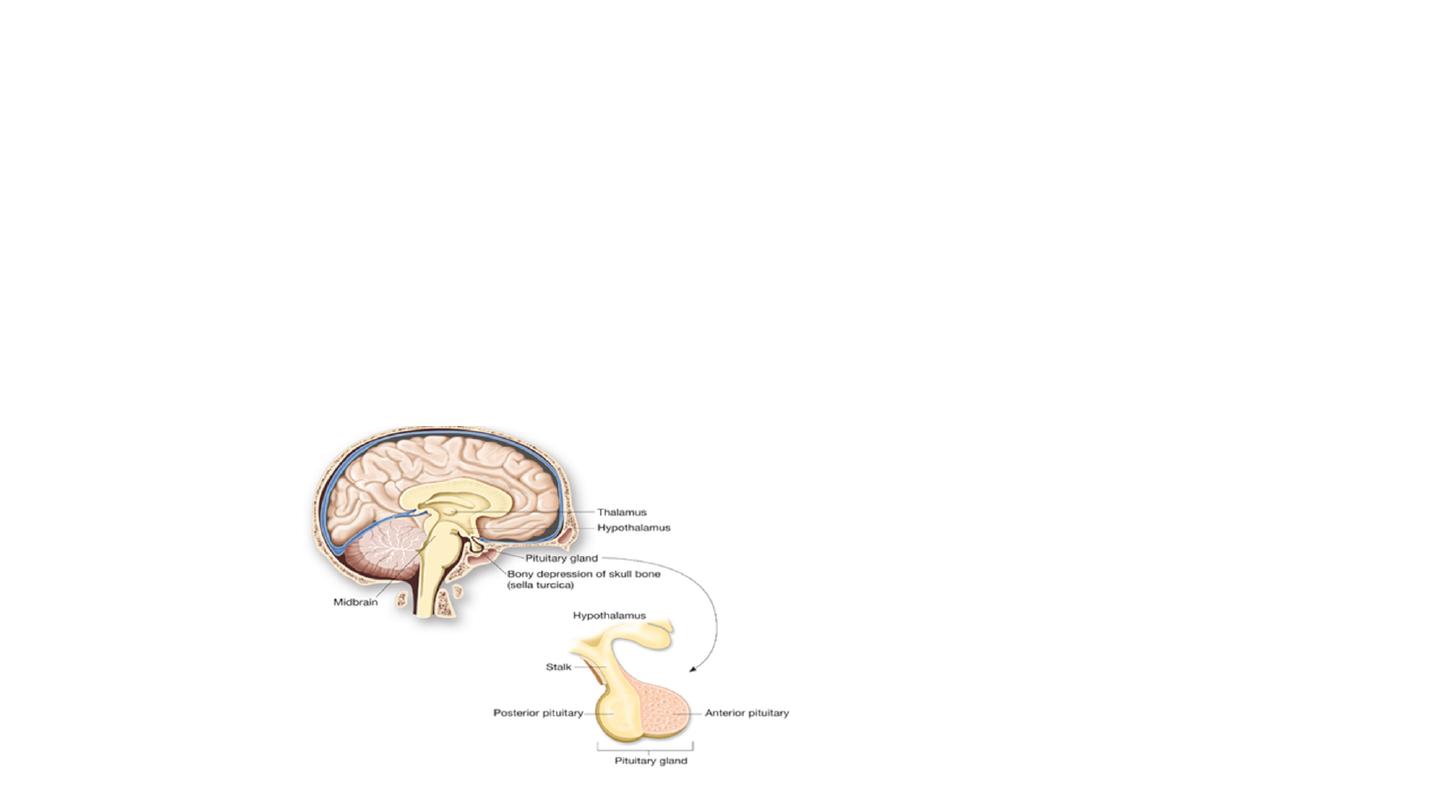
Pituitary gland:
It is an endocrine gland about the size of a pea
and weighing 0.5 grams in human. It is a protrusion off the bottom of the hypothalmus at the base
of the brain and is surrounded by a small bony
cavity
(sella turcica). The pituitary gland is attached to the hypothalmus by a stalk composed of
neuronal axons. The anterior and posterior lobes of the pituitary are functionally, anatomically, and embryologically distinct. Whereas the anterior
pituitary contains abundant hormone-secreting epithelial cells, the posterior pituitary is composed largely of unmyelinated secretory neurons. In
humans the intermediate lobe does not exist as a distinct anatomic structure.
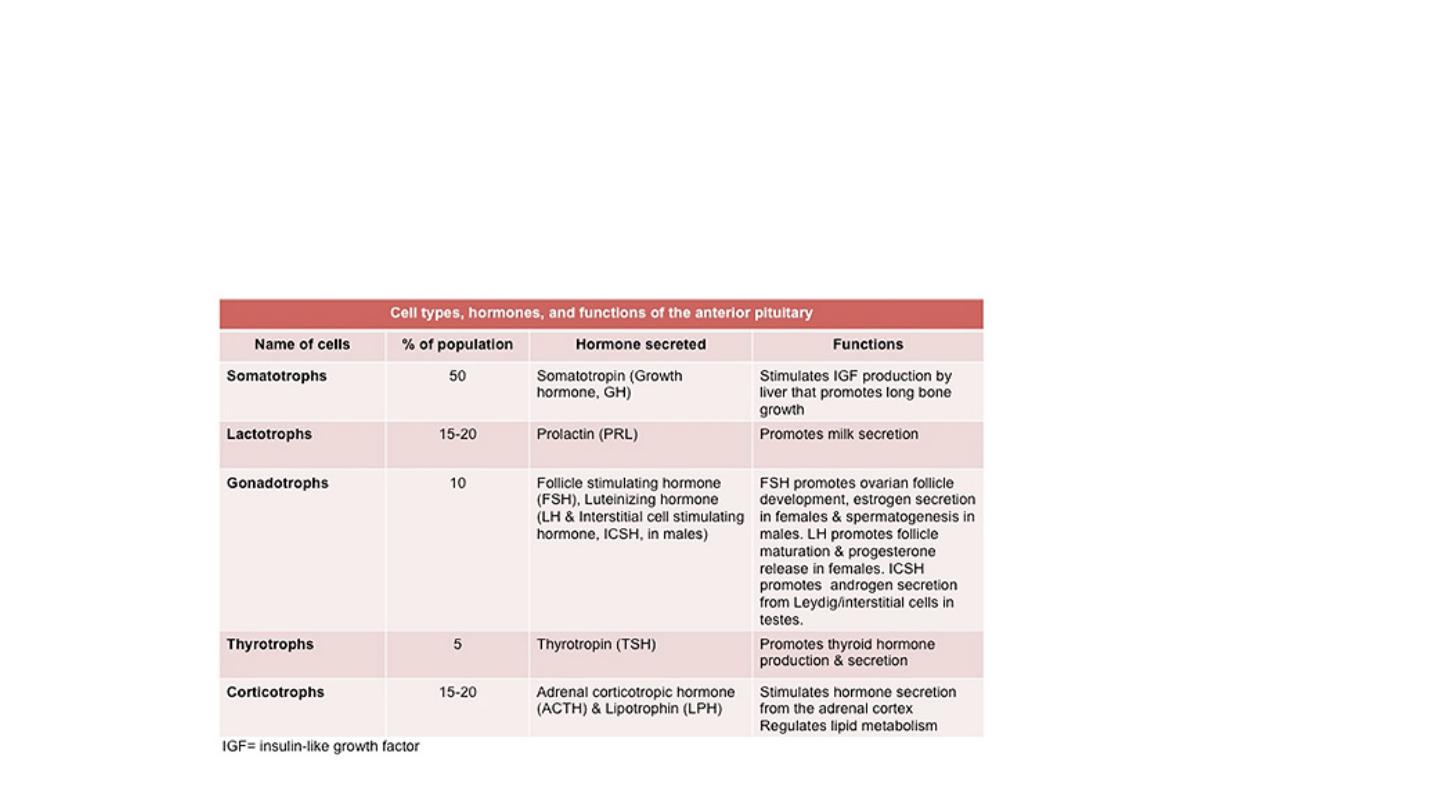
Anterior Pituitary:
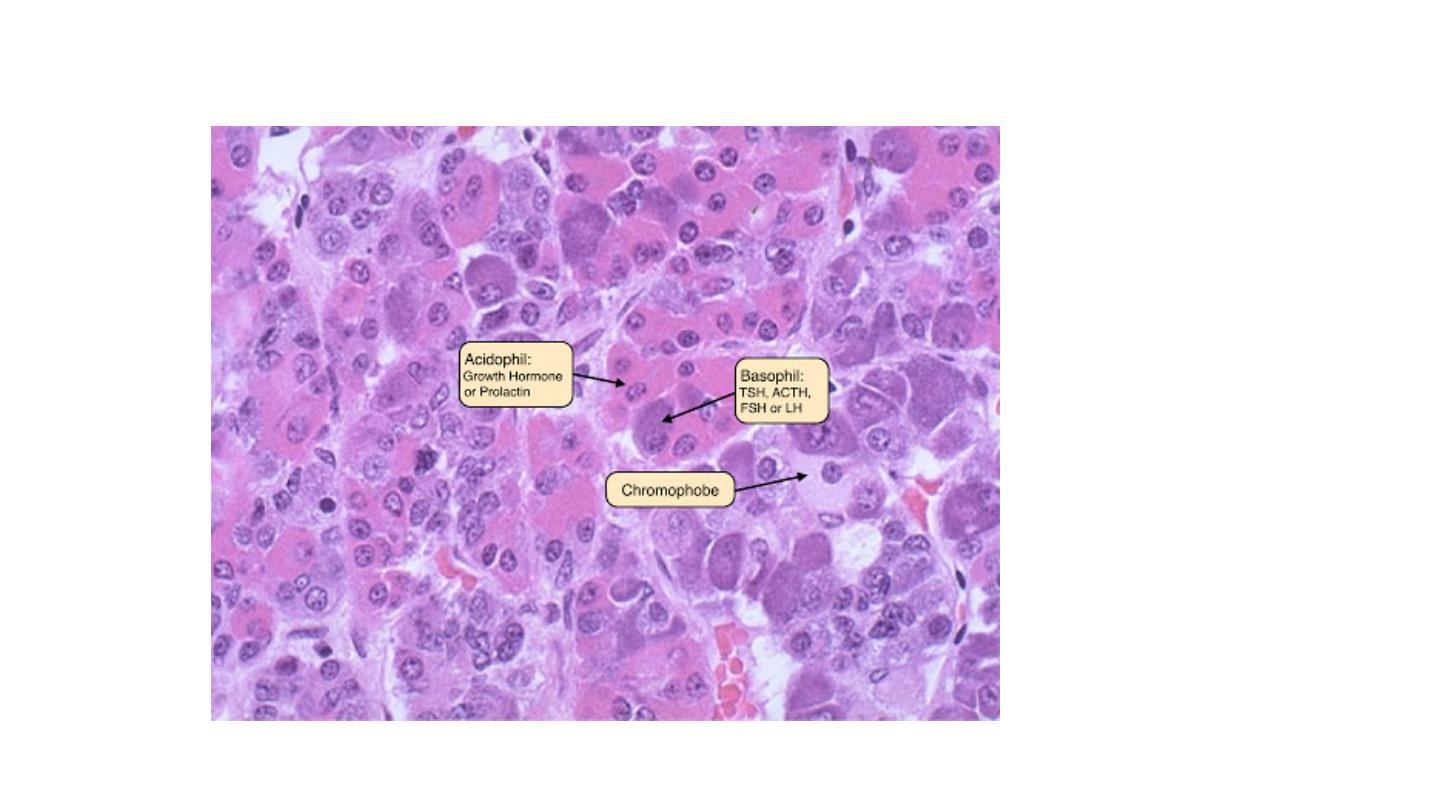
The cells of pituitary gland generally
divided
to three types depend on the colour of the cytoplasm. Basophilic cells, eosinophilic cells and poorly
staining (chromophobic cells). These cells have various
trophic
polypeptide hormones in their cytoplasm. The release of trophic hormones is
under the control of factors produced by in the hypothalamus; while most of hypothalamic factors are stimulatory and promote pituitary hormones
release, others like somatostatin and dopamine are inhibitory in their effects.
Posterior Pituitary:
It only secretes two hormones:
Antidiuretic hormone (ADH) which acts on the kidney, and oxytocin, which acts on the uterus.

Disease of Pituitary Glands:
Adenoma: is the most common cause of hyperpituitarism. It arises in the anterior lobe. The less common
causes include hyperplasia and carcinomas of the anterior pituitary,secretion of hormones by some
extrapituitary tumours, and certain hypothalamic disorders. Pituitary adenomas can be functional in which
the gland secrets the hormones in excess amount and that leads to clinical manifestations or could be non
functional in which the hormones produce at the tissue level only, without clinical manifestations. Non
functioning adenomas are come to clinical attention at later stage when become large in size, destroying the
adjacent anterior pituitary parenchyma and hypopituitarism features can result. Both types usually are
composed of single cell type and produce a single predominant hormone. Some pituitary adenomas can
secrete two different hormones (growth hormone and prolactin being the most common combination),
rarely, pituitary adenomas are plurihormonal. Most pituitary adenomas occur as sporadic and only 5% of
cases, adenomas occur as a result of an inherited predisposition.

Pathogenesis of Pituitary adenoma
Approximately40 % of sporadic growth hormone-secreting somatotroph cell adenomas and a minority
of adenocorticotrophic hormone (ACTH)-secreting corticotroph cell adenomas bear GNAS1 mutations.
Four genes have been identified as a cause of familial pituitary adenomas : MEN1(Multiple Endocrine
Neoplasia I), CDKN1B(Cyclin Dependent Kinase Inhibitor 1B) (P27), PRKAR1A (Protein Kinase,
CAMP-Dependent, Regulatory Subunit Type I Alpha ) and AIP(Aryl Hydrocarbon Receptor Interacting
Protein). Germline mutations of the men1 gene are responsible for multiple endocrine neoplasia
syndrome type 1. The product of the CDKN1Bgene is the cell cycle checkpoint regulator P27; germline
mutations of CDKN1B are responsible for a subset of patients with MEN-1 like syndrome who lack
MEN1 abnormalities. Patients with AIP germline mutations often develop GH-secreting adenomas at
younger age (before 35 years) than the typical for sporadic GH adenoma patients. Mutations of TP53 in
pituitary adenomas are associated with a propensity for aggressive behaviour such as invasion and
recurrence.

Morphology:
Pituitary adenomas can be diagnosed as microadenomas if they are less than 1 cm in diameter and
macroadenomas if they exceed 1 cm in diameter. The usual pituitary adenoma is a well-
circumscribed, soft lesion that may in case of smaller tumour be confined by sella turcica. Larger
lesions may compress the adjacent structures. In about 30% of cases, the adenomas are non
encapsulated and infiltrate adjacent bone, dura. Foci of hemorrhage and or necrosis are common in
larger adenomas.
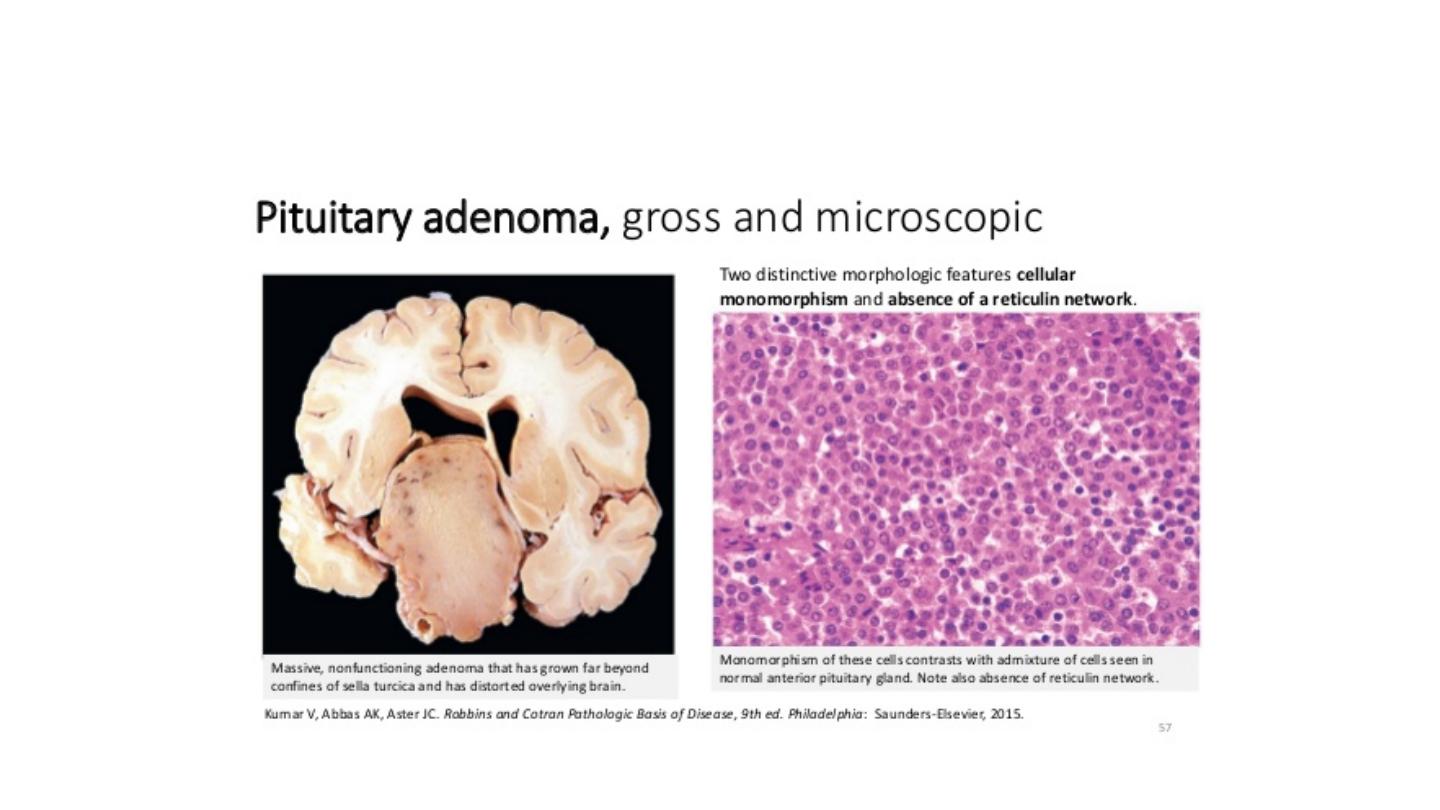
Pituitary adenomas are composed of relatively uniform, polygonal cells arrayed in sheets, cords, or
papillae. Supporting connective tissue in sparse accounting for the soft gelatinous consistency of
many of these tumours. The nuclei of the neoplastic cells may be uniform or pleomorphic. Mitotic
activity usually is scanty. The cytoplasm of the cells may be acidophilic, basophilic or chromophobic,
depending on the type and amount of secretory product within the cell, but it is fairly uniform
throughout the neoplasm. This cellular monomorphism and the absence of a significant reticulin
network distinguish pituitary adenomas from non-neoplastic anterior pituitary parenchyma.

Types of Pituitary Adenoma:
Prolactinomas: They are the most common type of hyper functioning pituitary adenoma.
They range in size from small microadenomas to large,expensile tumours associated with
considerable mass effect. Prolactin is demonstrable within the cytoplasm of neoplastic cells
by immunohistochemical techniques. Hyperprolactinemia causes amenorrhea, galactorrhea,
and infertility. Hyperprolactinemia may be caused by conditions or factors other than
prolactin-secreting pituitary adenomas, including pregnancy, high dose estrogen therapy,
renal failure, hypothyroidism, hypothelmic lesions and dopamine- inhibiting drug.

Adrenocorticotrophic Hormone-Producing (Corticotroph Cell Adenomas): Most corticotroph
cell adenomas are small (microadenomas) at the time of diagnosis. These adenomas stain
positively with periodic acid Schiff (PAS) stains, as a result of the accumulation of glycosylated
ACTH protein. As in the case of other pituitary hormones, the secretory granules can be detected
by immuohistochemical methods. Corticotroph cell adenomas may be clinically silent or may
cause hypercotisolism, manifested clinically as Cushing Syndrome, because the stimulatory effect
of ACTH on the adrenal cortex. Large, clinically aggressive corticotroph cell adenomas may
develop after surgical removal of the adrenal glands for treatment of Cushing Syndrome. In most
instances, this condition, known as Nelson syndrome, results from loss of the inhibitory effects of
adrenal corticosteroids on a pre-existing corticotroph microadenoma. Because the adrenals are
absent in persons with Nelson syndrome, hypercortisolism does not develop. Instead, patients
present with the mass effects of pituitary tumour. In addition, because ACTH is synthesised as
part of larger prohormone substance that includes melanocyte-stimulating hormone (MSH), hyper
pigmentation also may be a feature.

Growth Hormone-Producing (Somatotroph Cell) Adenomas
They constitute the second most common type of functional pituitary adenoma. Because
the clinical manifestations of excessive growth hormone may be subtle, somatotroph cell
maybe quite large by the time they come to clinical attention. On microscopic
examination, growth hormone-producing adenomas are composed of densely or sparsely
granulated cells, and immunohistochemical staining demonstrates growth hormone with in
the cytoplasm of the neoplastic cells. Small amounts of immunoreactive prolactin often
are present as well. Persistent hypersecretion of growth hormone stimulates the hepatic
secretion of insulin-like growth factor 1(somatomedin C), which causes many of the
clinical manifestations.
If a growth hormone-secreting adenoma occurs before the epiphysis close, excessive level
of growth hormone results in gigantism. If elevated level of growth hormone persist, or
develop after closure of the epiphyses, affected persons develop acromegaly. Growth
hormone excess also is associated with a number of other disturbances including abnormal
glucose tolerance and diabetes mellitus, generalized muscle weakness, hypertension,
arthritis, osteoporosis and congestive heart failure.

Hypopituitarism: Hypofunction of the anterior pituitary may occur with loss or absence of 75% or
more of the anterior pituitary parenchyma. This may be congenital (rare) or may result from wide
range of acquired abnormalities. Less frequently, disorder that interfere with the delivery of pituitary
hormones releasing factors from the hypothalamus like hypothalamic tumour may cause
hypofunction of pituitary gland.
Posterior Pituitary Syndromes: The clinically important posterior pituitary syndromes involve
ADH production. They include diabetes insipidus and secretion of inappropriately high levels of
ADH.

Thyroid gland:
The thyroid gland: is an endocrine gland in the neck, consisting of two lobes connected by an isthmus. It is found at the
front of the neck. The thyroid gland secretes three hormones, namely the thyroid hormones (thyroxine/T
4
and
triiodothyronine/T
3
) and calcitonin.
They collectively called thyroid hormones. The main hormone is thyroxine. Thyroid hormones act throughout the body,
influencing metabolism, growth and development, and body temperature. During infancy and childhood, adequate thyroid
hormone is crucial for brain development. The tissue of the thyroid gland is composed mostly of thyroid follicles. The
follicles are made up of a central cavity filled with a sticky fluid called colloid, surrounded by a wall of epithelial follicle
cells.
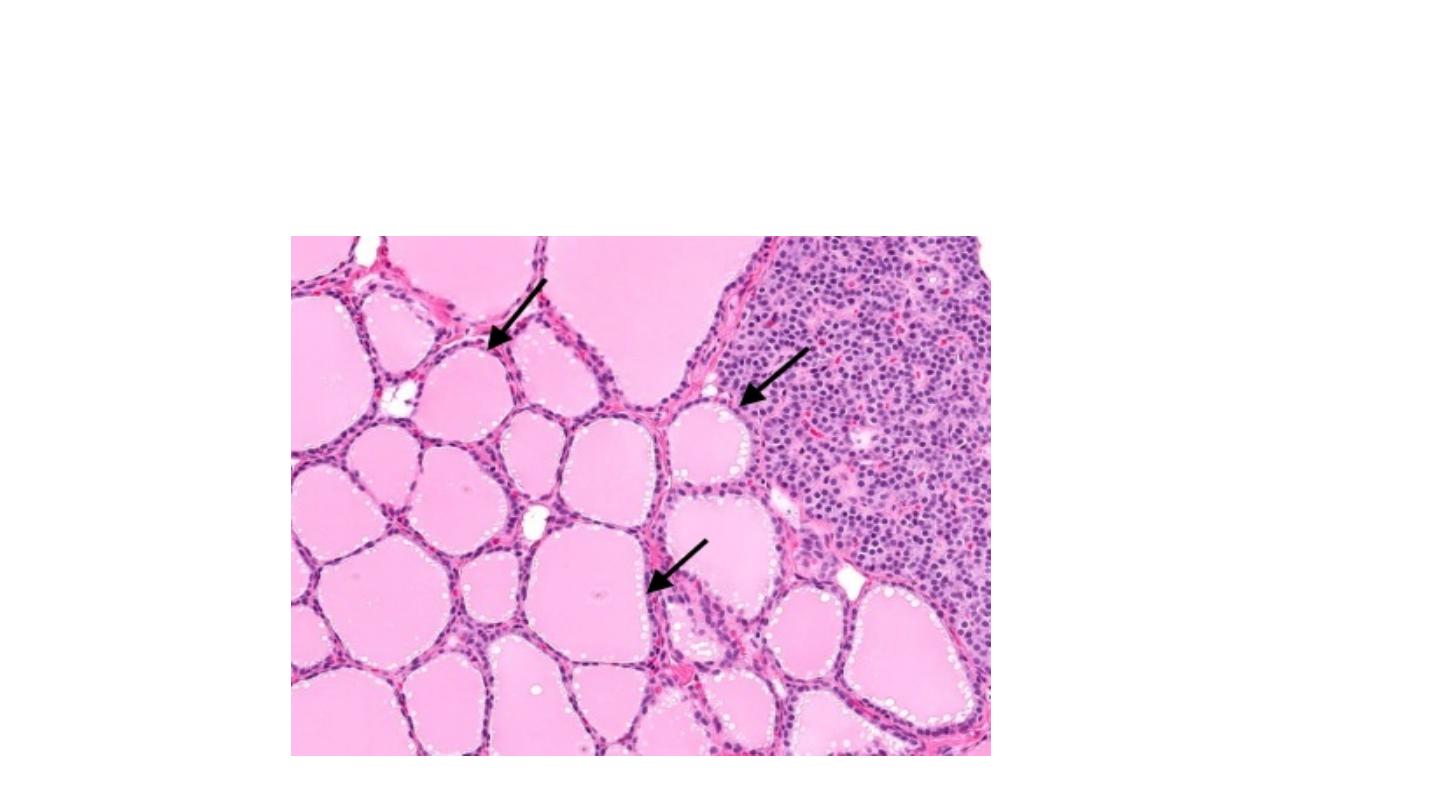
Figure: Histology of thyroid gland
Regulation of TH Synthesis
The release of T
3
and T
4
from the thyroid gland is regulated by thyroid-stimulating hormone (TSH).
Low blood levels of T
3
and T
4
stimulate the release of thyrotropin-releasing hormone (TRH) from the
hypothalamus, which triggers secretion of TSH from the anterior pituitary. In turn, TSH stimulates the
thyroid gland to secrete T
3
and T
4
. The levels of TRH, TSH are regulated by a negative feedback
system in which increasing levels of T
3
and T
4
decrease the production and secretion of TSH.
•
: A general term for thyroid swelling. Goiters can be harmless, or can represent iodine
deficiency or a condition associated with thyroid inflammation called Hashimoto’s thyroiditis.
•
: Inflammation of the thyroid, usually from a viral infection or autoimmune condition.
Thyroiditis can be painful, or have no symptoms
•
: Excessive thyroid hormone production. Hyperthyroidism is most often caused by
Graves disease or an overactive thyroid nodule.
•
: Low production of thyroid hormone. Thyroid damage caused by autoimmune disease
is the most common cause of hypothyroidism.

Thyroiditis: Thyroiditis, or inflammation of the thyroid gland, encompasses a
diverse group of disorders characterized by some form of thyroid inflammation.
These diseases include conditions that result in acute illness with severe thyroid
pain (e.g., infectious thyroiditis, subacute granulomatous thyroiditis) and
disorders in which there is relatively little inflammation and the illness is
manifested primarily by thyroid dysfunction (subacute lymphocytic thyroiditis
and fibrous thyroiditis). on the more common and clinically significant types of
thyroiditis: (1) Hashimoto thyroiditis (or chronic lymphocytic thyroiditis), (2)
subacute granulomatous thyroiditis, and (3) subacute lymphocytic thyroiditis.

HASHIMOTO THYROIDITIS
Hashimoto thyroiditis (or chronic lymphocytic thyroiditis) is the most common cause of
hypothyroidism in areas of the world where iodine levels are sufficient. It is characterized by
gradual thyroid failure because of autoimmune destruction of the thyroid gland. This disorder is
most prevalent between 45 and 65 years of age and is more common in women than in men, with a
female predominance of 10:1 to 20:1. Although it is primarily a disease of older women, it can occur
in children and is a major cause of non endemic goiter in children. Hashimoto thyroiditis comes to
clinical attention as painless enlargement of the thyroid, usually associated with some degree of
hypothyroidism, in a middle-aged woman.
The enlargement of the gland is usually symmetric and diffuse, but in some cases, it may be
sufficiently localized to raise a suspicion of neoplasm. In the usual clinical course,
hypothyroidism develops gradually. In some cases, however, it may be preceded by transient thyrotoxicosis
caused by disruption of thyroid follicles, with secondary release of thyroid hormones. During this phase, free T
4
and T
3
levels are elevated, TSH is diminished, and radioactive iodine uptake is decreased. As hypothyroidism
supervenes, T
4
and T
3
levels progressively fall, accompanied by a compensatory increase in TSH. Patients with
Hashimoto thyroiditis are at increased risk for developing other concomitant autoimmune diseases, both
endocrine (type 1 diabetes, autoimmune adrenalitis), and non endocrine (systemic lupus erythematosus,
myasthenia gravis, and Sjögren syndrome ), and also at increased risk for the development of B-cell non-
Hodgkin lymphomas. However, there is no established risk for developing thyroid epithelial neoplasms.

Pathogenesis.
Hashimoto thyroiditis is an autoimmune disease in which the immune system reacts
against a variety of thyroid antigens. The overriding feature of Hashimoto thyroiditis is
progressive depletion of thyroid epithelial cells (thyrocytes), which are gradually
replaced by mononuclear cell infiltration and fibrosis. Multiple immunologic
mechanisms may contribute to the death of thyrocytes. Sensitization of autoreactive
CD4+ T-helper cells to thyroid antigens appears to be the initiating event. The effectors
mechanisms for thyrocyte death include the following:
• CD8+ cytotoxic T cell-mediated cell death
• Cytokine-mediated cell death: CD4+ T cells produce inflammatory cytokines such as IFN-γ
with resultant recruitment and activation of macrophages and damage to follicles.
• Binding of antithyroid antibodies (anti-TSH receptor antibodies, antithyroglobulin, and
antithyroid peroxidase antibodies)

Morphology.
Macroscopic examination: The thyroid is often diffusely enlarged, although more
localized enlargement may be seen in some cases. The capsule is intact, and the gland is
well demarcated from adjacent structures. The cut surface is pale, yellow-tan, firm, and
somewhat nodular.
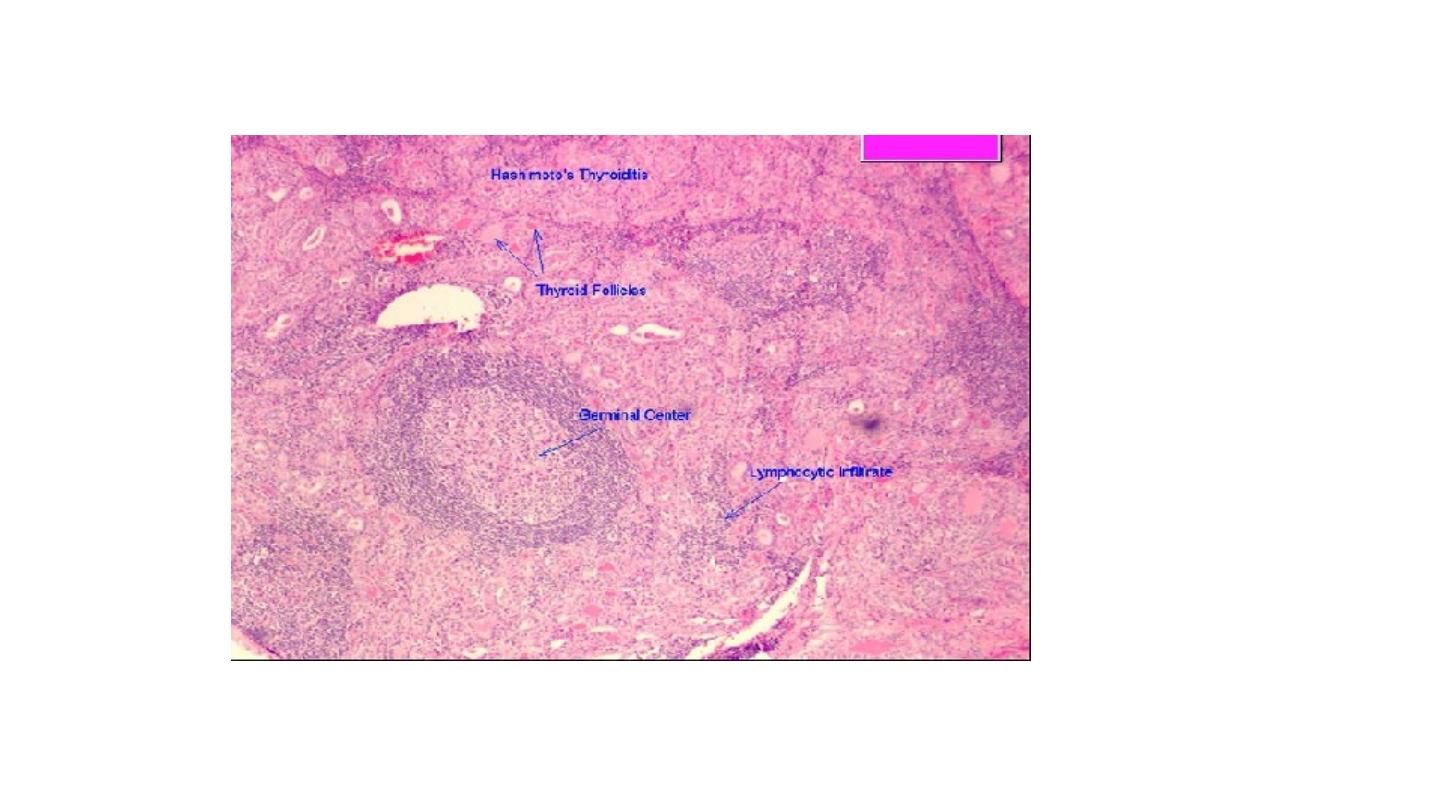
Microscopic examination reveals extensive infiltration of the parenchyma by a
mononuclear inflammatory infiltrate containing small lymphocytes, plasma cells, and
well-developed germinal centers. The thyroid follicles are atrophic and are lined in many
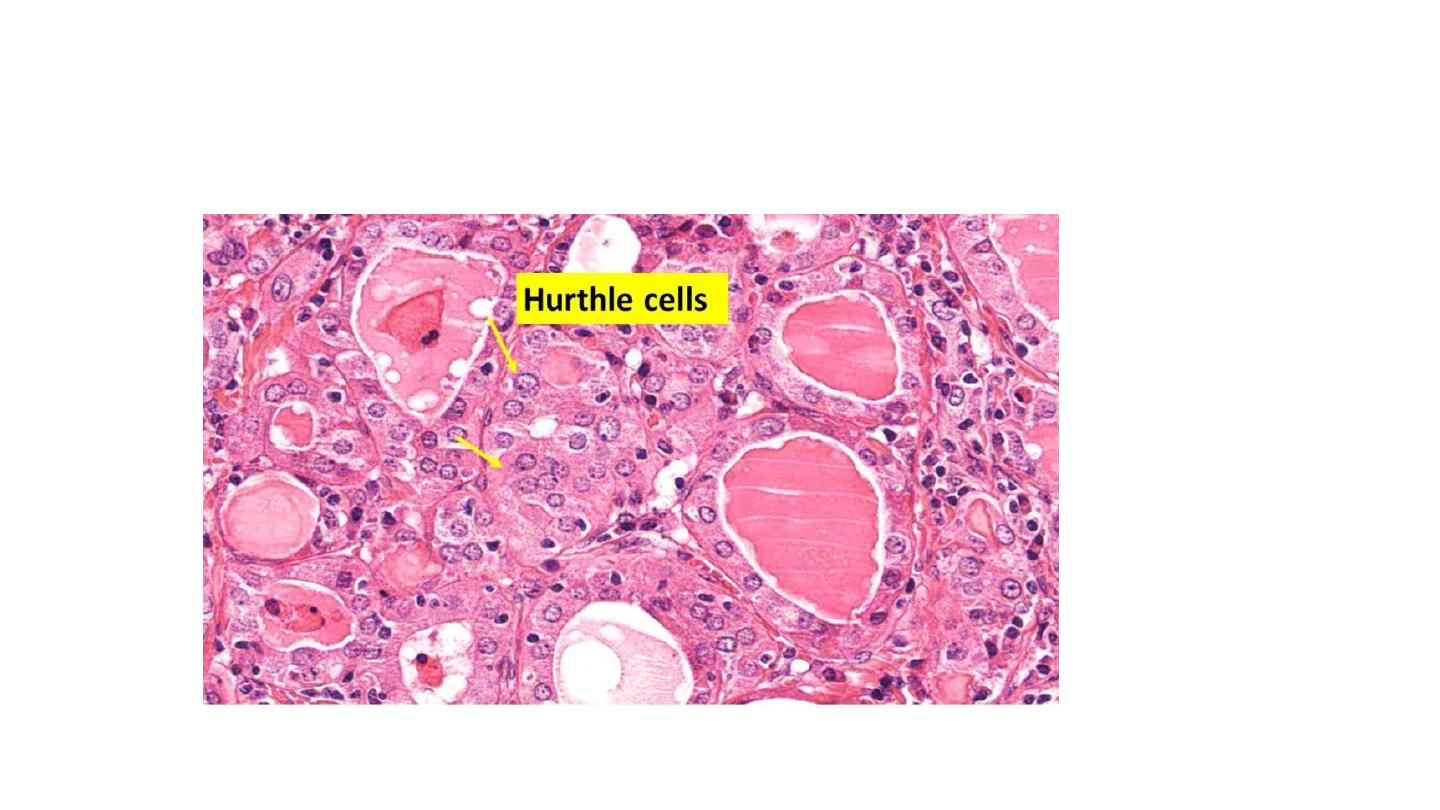
areas by epithelial cells distinguished by the presence of abundant eosinophilic, granular
cytoplasm, termed Hürthle cells. This is a metaplastic response of the normally low
cuboidal follicular epithelium to ongoing injury. In fine-needle aspiration biopsies, the
presence of Hürthle cells in conjunction with a heterogeneous population of lymphocytes is
characteristic of Hashimoto thyroiditis. In "classic" Hashimoto thyroiditis, interstitial
connective tissue is increased and may be abundant. A fibrous variant is characterized by
severe thyroid follicular atrophy and dense "keloid-like" fibrosis, with broad bands of
acellular collagen encompassing residual thyroid tissue. Unlike Reidel thyroiditis, the
fibrosis does not extend beyond the capsule of the gland.

SUBACUTE (GRANULOMATOUS) THYROIDITIS
Subacute thyroiditis, which is also referred to as granulomatous thyroiditis or De Quervain thyroiditis,
occurs much less frequently than does Hashimoto disease. The disorder is most common between
the ages of 30 and 50 and, like other forms of thyroiditis, affects women considerably more often than
men (3:1 to 5:1). The presentation of subacute thyroiditis may be sudden or gradual. It is characterized
by pain in the neck, which may radiate to the upper neck, jaw, throat, or ears, particularly when
swallowing. Fever, fatigue, malaise, anorexia, and myalgia accompany a variable enlargement of the
thyroid. The resultant thyroid inflammation and hyperthyroidism are transient, usually diminishing in 2
to 6 weeks, even if the patient is not treated. It may be followed by a period of transient, usually
asymptomatic hypothyroidism lasting from 2 to 8 weeks, but recovery is virtually always complete.
The transient hyperthyroidism, as in other cases of thyroiditis, is due to disruption of thyroid follicles
and release of excessive thyroid hormone. Nearly all patients have high serum T
4
and T
3
levels and low
serum TSH levels. Radioactive iodine uptake is low because of suppression of TSH. unlike in
hyperthyroid states such as Graves disease, radioactive iodine uptake is diminished. After recovery,
generally in 6 to 8 weeks, normal thyroid function returns.

Pathogenesis.
Subacute thyroiditis is believed to be caused by a viral infection or a post viral
inflammatory process. The majority of patients have a history of an upper
respiratory infection just before the onset of thyroiditis. The disease has a seasonal
incidence, with occurrences peaking in the summer, and clusters of cases have been
reported in association with coxsackie virus, mumps, measles, adenovirus, and other
viral illnesses. Although the pathogenesis of the disease is unclear, one model
suggests that it results from a viral infection that provides an antigen, either viral or
a thyroid antigen that is released secondary to virus-induced host tissue damage.
This antigen stimulates cytotoxic T lymphocytes, which then damage thyroid
follicular cells. In contrast to autoimmune thyroid disease, the immune response is
virus-initiated and not self-perpetuating, so the process is limited.

Morphology.
The gland may be unilaterally or bilaterally enlarged and firm, with an intact capsule. It
may be slightly adherent to surrounding structures. On cut section, the involved areas are
firm and yellow-white and stand out from the more rubbery, normal brown thyroid
substance.
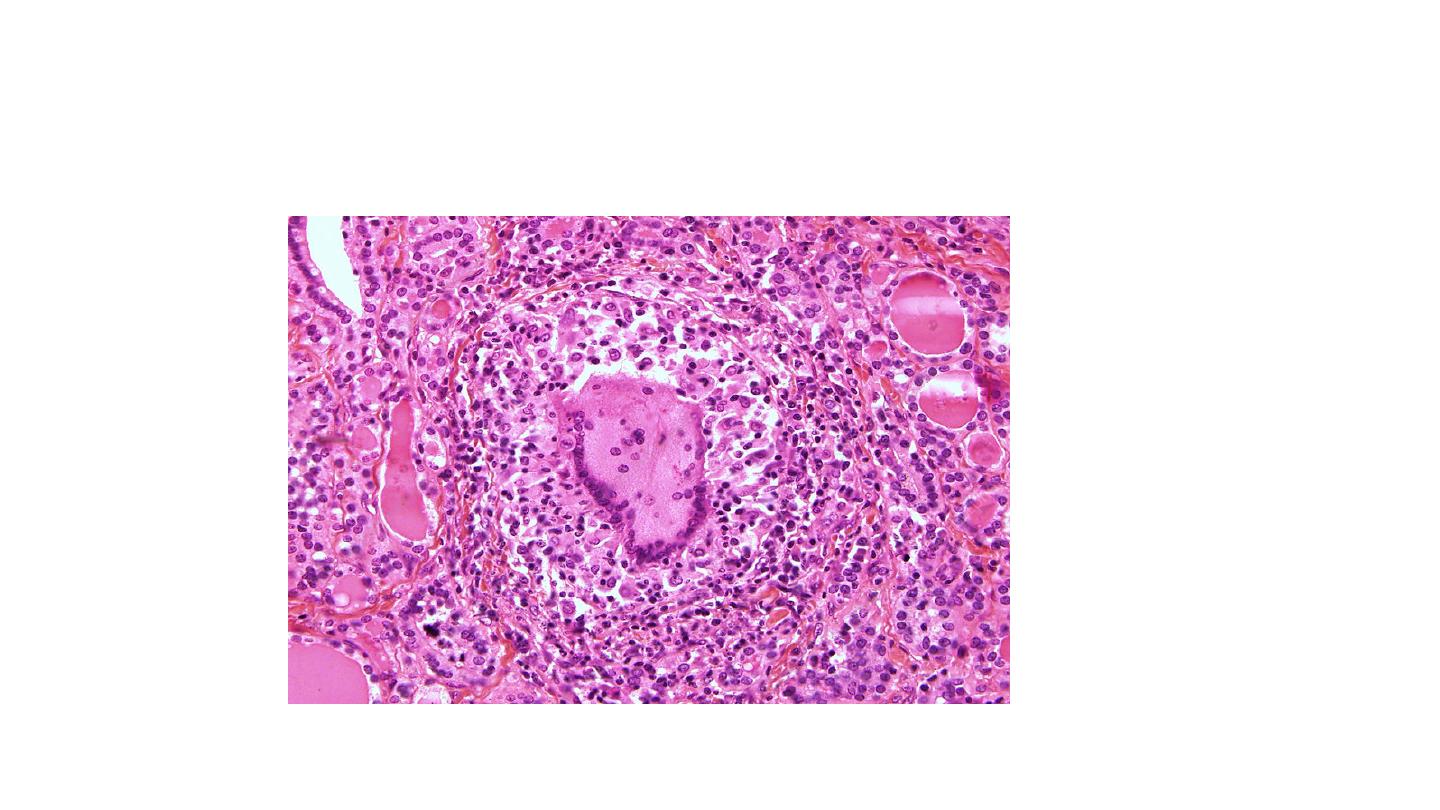
Histologically, the changes are patchy and depend on the stage of the disease. Early in the
active inflammatory phase, scattered follicles may be entirely disrupted and replaced by
neutrophils forming micro abscesses. Later, the more characteristic features appear in the
form of aggregations of lymphocytes, and plasma cells about collapsed and damaged
thyroid follicles. Multinucleate giant cells enclose naked pools or fragments of colloid),
hence the designation granulomatous thyroiditis. In later stages of the disease, a chronic
inflammatory infiltrate and fibrosis may replace the foci of injury. Different histologic
stages are sometimes found in the same gland, suggesting waves of destruction over a
period of time.
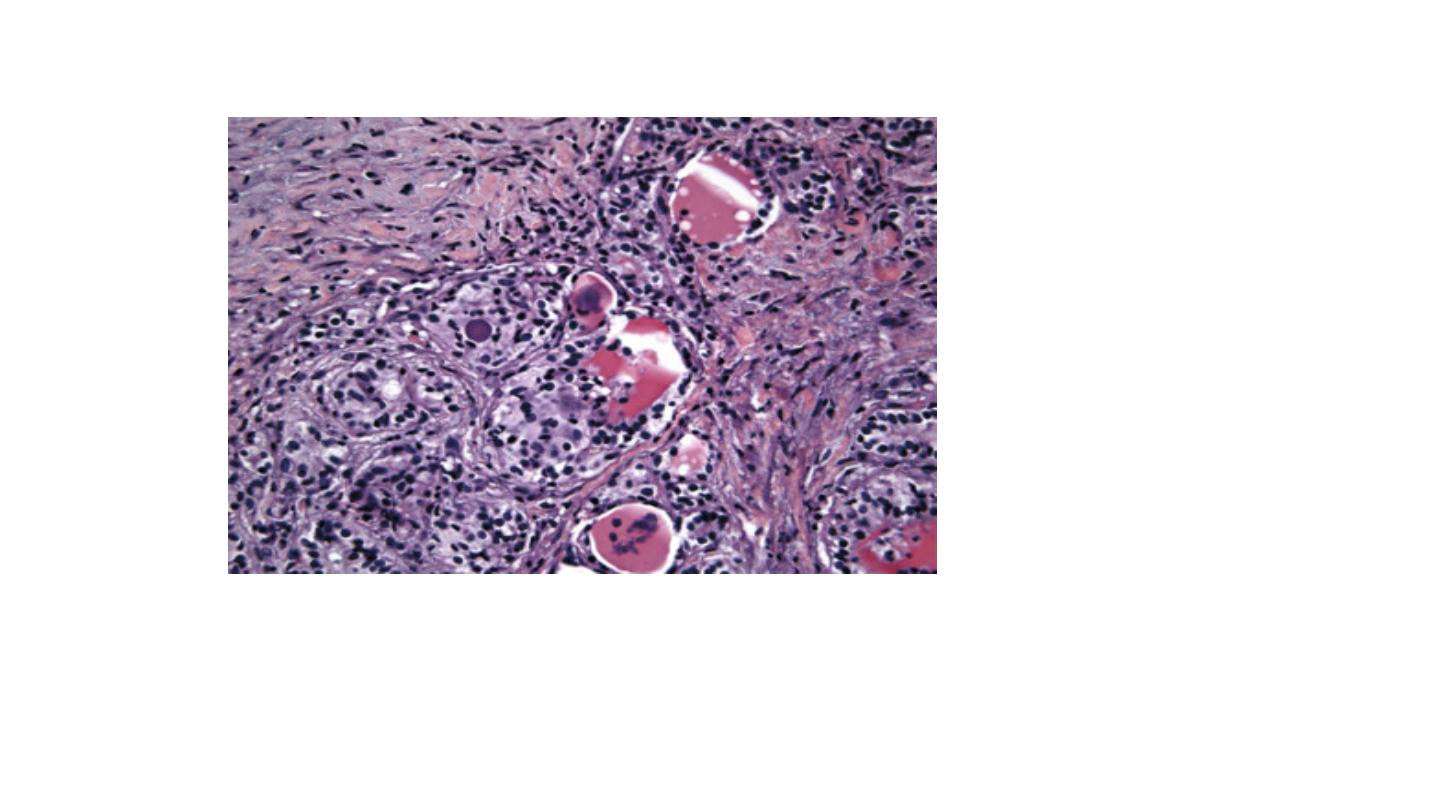
Subacute (viral or postviral) thyroiditis. Diffuse neutrophilic invasion with active
destruction of follicles and a multinucleated giant cell. Fibrosis and near-complete loss
of follicles have occurred.

SUBACUTE LYMPHOCYTIC (PAINLESS) THYROIDITIS
Subacute lymphocytic thyroiditis, which is also referred to as painless thyroiditis or silent
thyroiditis, is an uncommon cause of hyperthyroidism. It usually comes to clinical attention
because of mild hyperthyroidism, goitrous enlargement of the gland, or both. Although it can
occur at any age, it is most often seen in middle-aged adults and is more common in women,
especially during the postpartum period (postpartum thyroiditis), than in men. Depending on
the study, the frequency of this form of thyroiditis varies considerably, from 1% to about 10%
of cases of hyperthyroid patients. The pathogenesis of this disorder is unknown. An
autoimmune basis has been suggested because some patients have elevated levels of
antibodies to thyroglobulin and thyroid peroxidase or a family history of thyroid
autoimmune disease.

The principal clinical manifestation of painless thyroiditis is hyperthyroidism. The patient may
have any of the common findings of hyperthyroidism (e.g., palpitations, tachycardia, tremor,
weakness, and fatigue). The thyroid gland is not usually tender but is minimally and diffusely
enlarged. Infiltrative ophthalmopathy and other manifestations of Graves disease are not
present. Patients with one episode of postpartum thyroiditis are at an increased risk of
recurrence following subsequent pregnancies. A minority of affected individuals eventually
progress to hypothyroidism. Some patients have no signs or symptoms, and the disorder is
detected incidentally during routine thyroid testing. Laboratory findings during periods of
thyrotoxicosis include elevated levels of T
4
and T
3
and depressed levels of TSH.

Morphology.
The thyroid appears normal on gross inspection. The most specific histologic features
consist of lymphocytic infiltration with hyperplastic germinal centers within the thyroid
parenchyma and patch disruption and collapse of thyroid follicles. Unlike in Hashimoto
thyroiditis, fibrosis and Hürthle cell metaplasia are not commonly seen.
Riedel thyroiditis: a rare disorder of unknown etiology characterized by extensive fibrosis
involving the thyroid and contiguous neck structures. The presence of a hard and fixed
thyroid mass clinically simulates a thyroid carcinoma. It may be associated with
idiopathic fibrosis in other sites in the body, such as the retroperitoneum. The presence
of circulating antithyroid antibodies in most patients suggests an autoimmune etiology.
