Clinical immunologyL-4
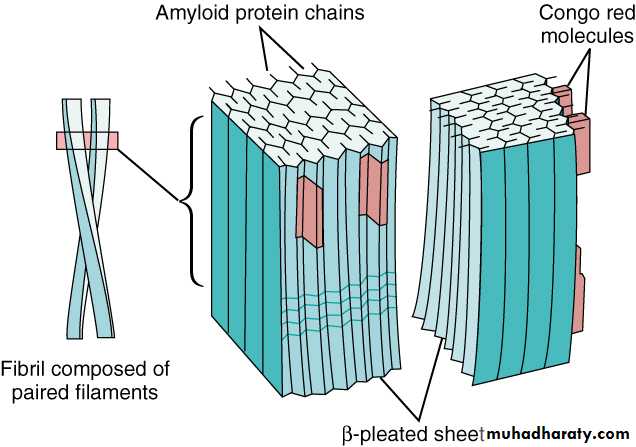
AmyloidosisIs a disorder of protein metabolism in which there is an extracellular deposition of pathological insoluble fibrillar proteins in organs and tissues. Characteristically, the amyloid protein consist of B-pleated sheets that are responsible for its insolubility and resistance to proteolysis.
.
Amyloidosis can be acquired or inherited. Classification is based on the nature of precursor of plasma protein( at least 20) that form the fibrillar deposit. The process for the production of the fibrils appears to be multifactorial and differs amongst the various types of amyloid.
This disease was named by Virchow in 1854 on the basis of color after staining with iodine &sulfuric acid.
All amyloid proteins share a unique fibrillar ultrastructure.
amyloid proteins can be deposited locally or can involve virtually every organ system of the body.Amyloid fibril deposition may have no clinical consequences or may lead to severe pathophysiological changes. Often the disease falls between these two extremes.
Regardless the etiology, the clinical diagnosis of amyloidosis is usually not made until the disease is far advanced.
The clinical manifestations of amyloidosis depends on the organ(s) affected.
The diagnosis of amyloidosis should be considered in all cases of unexplained nephrotic syndrome, cardiomyopathy, &peripheral neuropathy.CLASSIFICATION
Amyloid diseases are classified by etiology and type of protein deposited.1- acquired systemic amyloidosis
a- reactive(AA) (secondary) amyloidosis!
These are due to amyloid formed from serum amyloid A(SAA), which is an acute phase protein. It is, therefore, related to chronic inflammatory disorders and chronic infections.Clinical features depend on the nature of underlying disorder. Chronic inflammatory disorders include rheumatoid arthritis, inflammatory bowel disease and untreated familial Mediterranean fever.
In developing countries it is still associated with infectious diseases such as tuberculosis, bronchiectasis and osteomyelitis. AA amyloidosis often presents with chronic kidney disease, with hepatomegaly and splenomegaly. Macroglossia is not a feature and cardiac involvement is rare.
The degree of renal failure correlates with SAA level in a more favourable out come in patients with low normal levels.
b-light chain amyloid. (AL, primary) seen in patients with Myeloma and plasmacytoma.
It manifested as restrictive cardiomyopathy&
peripheral neuropathy.
c- Dialysis related amyloidosis:
This is due to B2- microglobulin producing amyloid fibrils in chronic dialysis patients. It frequently presents with carpal tunnel syndrome. It occurs 5-10 years after dialysis.dialysis associated(AB2M) amyloidosis.
Occurs 5-10 years of dialysisd- senile systemic amyloidosis:
Usually asymptomatic& occurs in patients over the age of 70 years.2-Familial(hereditary)amyloidosis: these are autosomally dominant transmitted diseases where the mutant protein forms amyloid fibrils, starting usually in the middle age.
.
hereditary systemic amyloidosis
An autosomal dominant disorder& manifested asperipheral& autonomic neuropathy& cardiomyopathy.
Examples include disorders such as familial amyloidosis polyneuropathy (FAP), cardiomyopathy or the nephrotic syndrome. Major foci of FAP occur in Portugal, japan and Sweden.
Local amyloidosis:
Deposits of amyloid fibril of various types can be localized to various organs or tissues( skin, heart or brain).
The brain is a common site of amyloid deposition.
Intracerebral and cerebrovascular amyloid deposits are seen in Alzheimer’s diseaseDiagnosis
the diagnosis is established by biopsy which may be of an affected organ , rectum or subcutaneous fat.The pathognomonic histological feature is apple-green birefringence of amyloid deposits when stained with Congo red dye and viewed under polarized light.
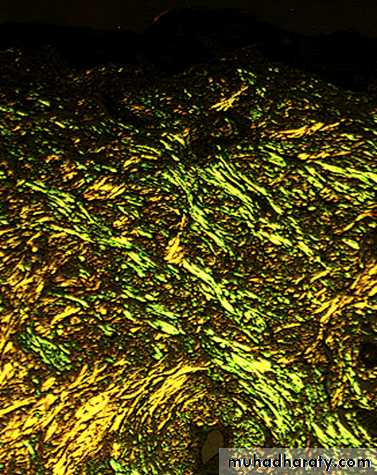
MANAGEMENT
the aims of treatment are to support the function of the affected organ and in acquired amyloidosis to prevent further amyloid deposition through treatment of the primary cause.Genetic counseling is an important aspect of treatment in heredofamilial amyloidosis.
Liver transplantation had been carried out since 1990 for patients with familial amyloid- polyneuropathy(FAP).Recent trials have indicated that a prednisolone/melphalan/
Colchicine's program can prolongs the life.
Autoimmune diseases
• Result from a failure of self-tolerance• Immunological tolerance is specific unresponsiveness to an antigen
• All individuals are tolerant of their own (self) antigens
these disorders are chronic and usually irreversible
incidence: 5%-7% of population, higher frequencies in women, increases with ageImmunological tolerance
This is the process by which the immune system distinguishes self from foreign tissues.Central tolerance occurs during lymphocyte development in the thymus and bone marrow.
T&B lymphocytes that recognize self antigens are deleted before they develop into fully immunocompetent cells.Some autoreactive cells
inevitably evade deletion &escaping to the peripheral circulation. These cells are controlled through peripheral tolerance mechanisms.These include the suppression of autoreactive cells by 'regulatory' T-cells& the generation of the hyporesponsivness 'anergy‘ in lymphocytes.
Failure of any of these
tolerance mechanisms may result in the development of autoimmune disease.Factors predisposing to autoimmune disease
Both genetic and environmental factors contribute to the development of autoimmune disease.The most important genetic determinants of autoimmune susceptibility are the HLA genes,
reflecting there importance in shaping of lymphocyte responses.Several environmental factors can trigger autoimmunity in genetically susceptible individuals.
The most widely studied of these is infection, as occurs in acute rheumatic fever fallowing streptococcal infection or reactive arthritis fallowing bacterial
infection.
A number of mechanisms have been postulated including cross- reactivity between the infectious pathogen and self determinants(molecular mimicry),
and release of sequestered antigens
which are not usually visible to the immune system from damaged tissue.Occasionally the development of autoimmune disease is a side effect of drug treatment, for example:
the metabolic products of the anesthetic drug halothane bind to liver enzymes, resulting in a structurally novel protein.
This is recognized as new(foreign)
antigen by the immune system causing hepatic necrosis.HLA-association in autoimmune disease
Disease HLA-ASSOCIATIONCLASSIFICATION OF AUTOIMMUNE DISEASE
TYPE DISEASEOrgan specific
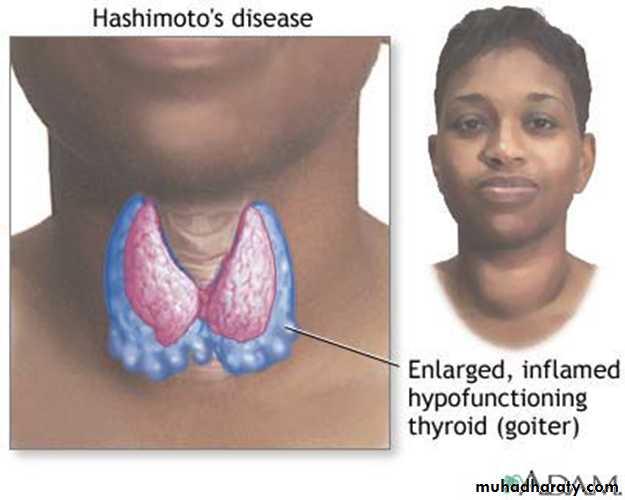
Immune response Grave's disease
Directed against Addison's dis.
Localized antigens Pernicious
anemia
Type-1 diabetes
Pemphigus vulgaris
Idiopathic thrombocytopenic purpura
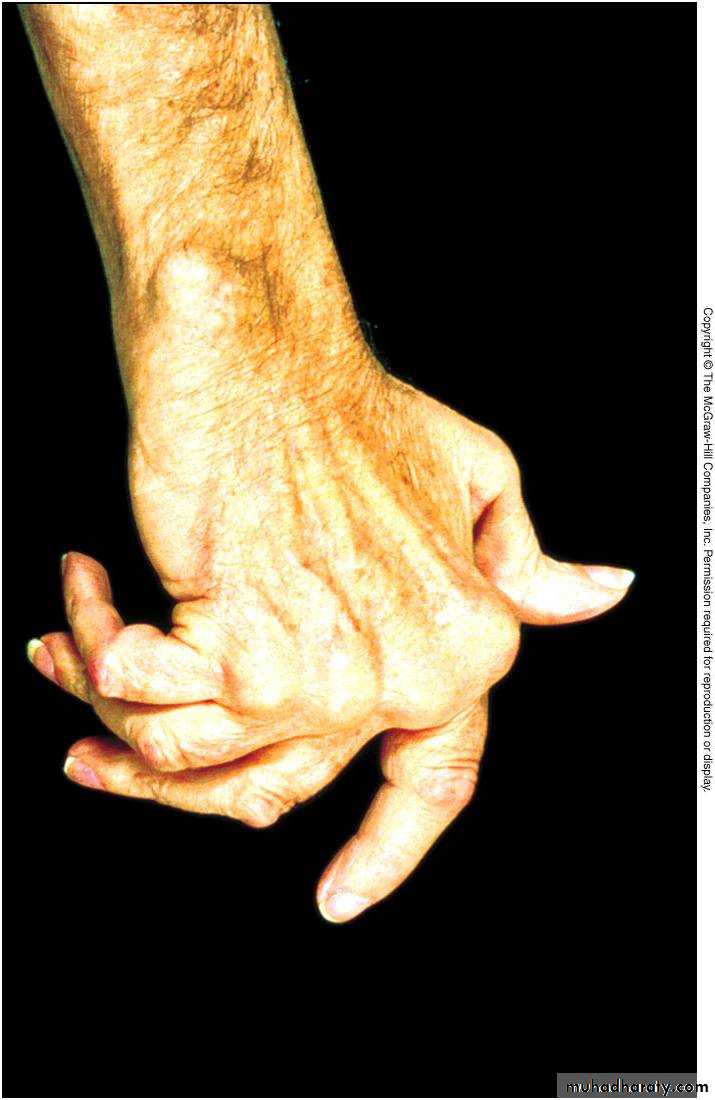
Autoimmune hemolytic anemia Myasthenia gravis Rheumatoid arthritis Dermatomyositis primary biliary cirrhosismultisystem
Immune response Systemic sclerosis
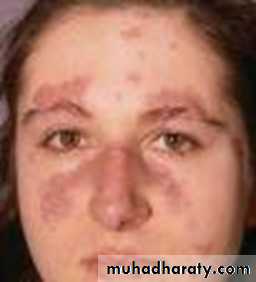
directed to Mixed connective widespread tissue disease target antigens SLE
*******************
Treatment of autoimmune diseases
Treatment aimed at:Killing dividing cells
Immunosuppressant
Controlling T cell signaling
Cyclosporin
Anti-inflammatory medications
Cortisone-like steroids
Replacement therapy
Insulin