Biochemistry
2nd stageDr.Lamees Majid Al-Janabi
Biosynthesis of Nonessential Amino Acids
A number of a.as can be synthesized from components of glycolytic pathway and the CAC . In addition several of the a.as can be formed from other a.as. These a.as called nonessential a.as i.e. they can be made available to the cells even though they are not included in the diet.
A. Synthesis from α-keto acids
Alanine, aspartate, and glutamate are synthesized by transfer of an amino group to the α-keto acids pyruvate, oxaloacetate, and α-ketoglutarate, respectively. These transamination reactions are the most direct of the biosynthetic pathways. Glutamate is unusual in that it can also be synthesized by the reverse of oxidative deamination, catalyzed by glutamate dehydrogenase .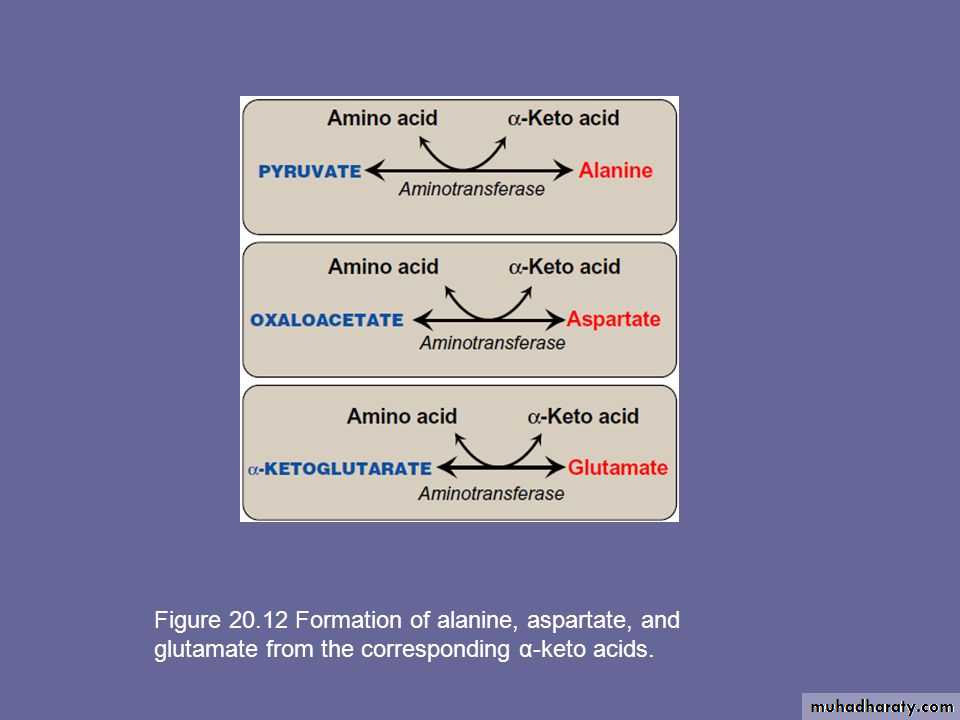
B. Synthesis by amidation
- Glutamine: Glutamate is formed by transamination when the above reaction is reversed. Glutamate also formed from alpha- ketoglutarate by glutamate dehydrogenase reaction.Aspartate & glutamate also formed as a result of the hydrolysis asparagine & glutamine by the enzyme Asparaginase & Glutaminase
Glutamine is synthesized as follow
glutamine synthetase
glutamate + ATP + NHз →→→→→→→→ glutamine + ADP + Pi
Asparagine:
Asparagine synthetase
aspartate +glutamine + ATP →→→→→ Asparagine + ADP + Pi
C. Proline
Proline can be formed from glutamate.
D. Serine, Glycine, Cysteine:
Serine is formed from 3 – phospho - glycerate (glycolytic intermediate)Glycine is generated by a single carbon transfer from serine to THFA
Cysteine can be synthesized from the carbon skeleton of serine.
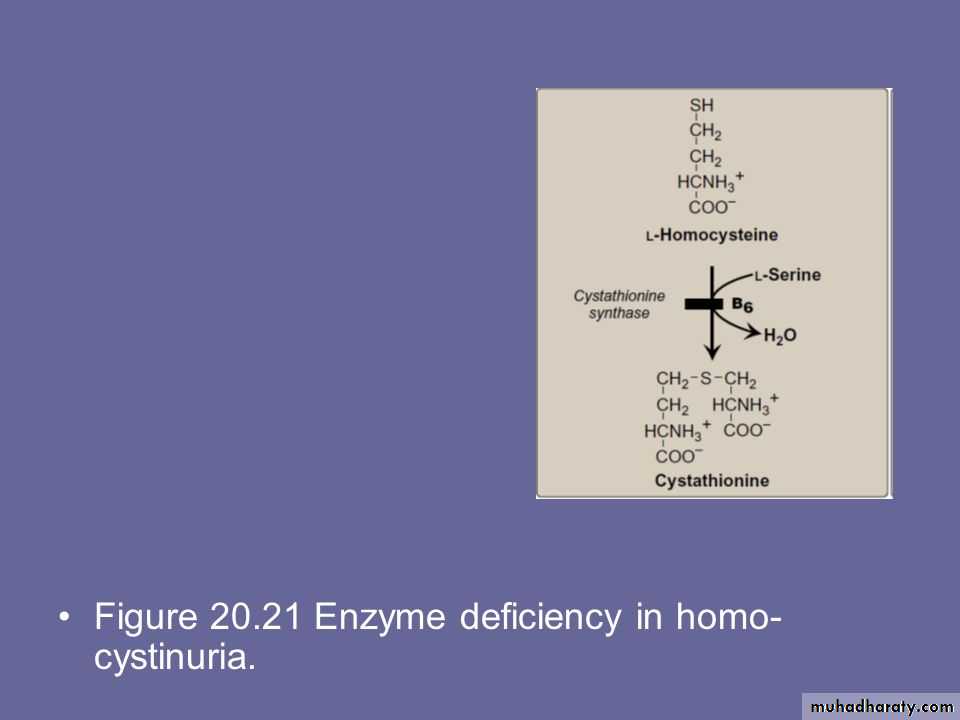
Cystathionine Synthase
homocysteine + serine cystathionineCystathionase
Cystathionine cysteine + alpha – ketobutyrateE. Tyrosine is formed from phenylalanine & oxygen by the NADPH dependent enzyme called phenylalanine hydroxylase which require the coenzyme tetrahydrobiopterin
phenylalanine + NADPH + H+ + O2→→ tyrosine + NADP + H2O
Tyrosine and cysteine are formed from an essential amino acid and, is therefore, nonessential only in the presence of adequate dietary phenylalanine and methionine.
Metabolic Defects in Amino Acid Metabolism
Inborn errors of metabolism are commonly caused by mutant genes that generally result in abnormal proteins, most often enzymes.
A. Phenylketonuria
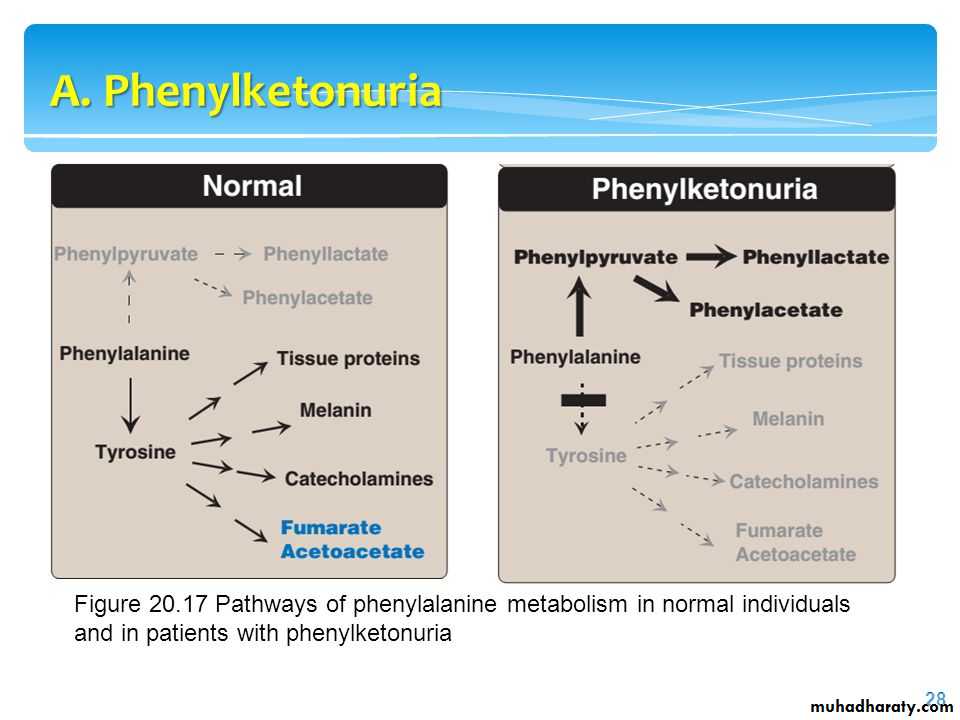
Phenylketonuria (PKU), caused by a deficiency of phenylalanine hydroxylase , PKU is the most common clinically encountered inborn error of amino acid metabolism (prevalence 1:15,000). Biochemically, it is characterized by accumulation of phenylalanine (and a deficiency of tyrosine).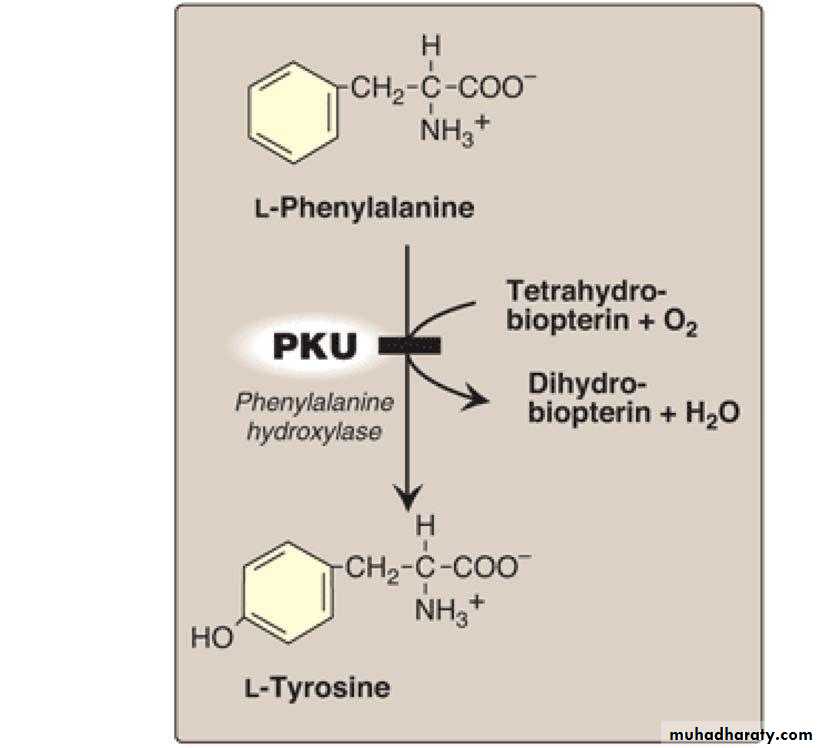
Characteristics of classic PKU:
Elevated phenylalanine: Phenylalanine is present in elevated concentrations in tissues, plasma, and urine.CNS symptoms: Mental retardation, failure to walk or talk, seizures, hyperactivity, tremor, microcephaly, and failure to grow are characteristic findings in PKU.
Hypopigmentation: Patients with phenylketonuria often show a deficiency of pigmentation (fair hair, light skin color, and blue eyes). The hydroxylation of tyrosine by tyrosinase, which is the first step in the formation of the pigment melanin, is competitively inhibited by the high levels of phenylalanine present in PKU.
B. Maple syrup urine disease
Maple syrup urine disease (MSUD) is a rare (1:185,000), autosomal recessive disorder in which there is a partial or complete deficiency in branched-chain α-keto acid dehydrogenase, an enzyme complex that decarboxylates leucine, isoleucine, and valine .These amino acids and their corresponding α-keto acids accumulate in the blood, causing a toxic effect that interferes with brain functions. The disease is characterized by feeding problems, vomiting, dehydration, severe metabolic acidosis, and a characteristic maple syrup odor to the urine. If untreated, the disease leads to mental retardation, physical disabilities, and even death.
C. Albinism
Albinism refers to a group of conditions in which a defect in tyrosine metabolism results in a deficiency in the production of melanin. These defects result in the partial or full absence of pigment from the skin, hair, and eyes. In addition to hypopigmentation, affected individuals have vision defects and photophobia (sunlight hurts their eyes). They are at increased risk for skin cancer.
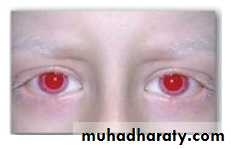
Patient with oculocutaneous albinism, showing white eyebrows and lashes.
D. HomocystinuriaThe homocystinurias are a group of disorders involving defects in the metabolism of homocysteine. The diseases are inherited as autosomal recessive illnesses, characterized by high plasma and urinary levels of homocysteine and methionine and low levels of cysteine.
The most common cause of homocystinuria is a defect in the enzyme cystathionine β-synthase, which converts homocysteine to cystathionine. Individuals who are homozygous for cystathionine β-synthase deficiency exhibit ectopia lentis (displacement of the lens of the eye), skeletal abnormalities, premature arterial disease, osteoporosis, and mental retardation.
E. Alkaptonuria
Alkaptonuria is a rare metabolic disease involving a deficiency in homogentisic acid oxidase, resulting in the accumulation of homogentisic acid. [Note: This reaction occurs in the degradative pathway of tyrosine .] The illness has three characteristic symptoms: homogentisic aciduria (the patient's urine contains elevated levels of homogentisic acid, which is oxidized to a dark pigment on standing , large joint arthritis, and black ochronotic pigmentation of cartilage and collagenous tissue . Patients with alkaptonuria are usually asymptomatic until about age 40.