Bioavailability of drugsBioavailability is the fraction of the dose of a drug contained in any dosage form that reaches the systemic circulation in unchanged or active form administered through any routeBioavailability = AUC (oral)/ AUC (I/V) x 100 (AUC is the area under the curve)When the drug is given orally, only part of the administered dose appears in the plasma. The area under the curve (AUC) can be measured by plotting plasma concentrations of the drug versus time.Drugs injected using intravenous route of administration have 100% bioavailability, while others have much less bioavailability, because:Not all the drug adsorbed Metabolism of the drug might occur before reaching circulation
Distribution of drugs:Penetration of a drug to the sites of action through the walls of blood vessels from the administered site after absorption is called drug distribution. Drugs distribute through various body fluid compartments.1-Plasma compartment: If a drug has a very large molecular weight or bindsextensively to plasma proteins, it is trapped within the plasma (vascular)compartment.2- Extracellular fluid: If a drug has a low molecular weight but is hydrophilic,it can move through the endothelium of the capillaries into the interstitial fluid.3- Total body water: If a drug has a low molecular weight and is hydrophobic,not only can it move into the interstitium through the slit junctions, but it canalso move through the cell membranes into the intracellular fluid.4- Other sites: In pregnancy, the fetus may take up drugs and thus increase the volume of distribution.
Volume of DistributionVolume of distribution (Vd) is the ratio between the amount of drug in body (dose given) (D) and the concentration of the drug (C) measured in blood or plasma. D Vd = ------ C
Factors affected distribution of drugs:
1. Protein binding of drug:Absorbed drug may become reversibly bound to plasma proteins (mainly albumin and to a less degree globulin). The active concentration of the drug is that part which is not bound, because only this fraction is free to leave the plasma. (a) Free drug leave plasma to site of action (b) binding of drugs to plasma proteins assists absorption (c) protein binding acts as a temporary store of a drug and tends to prevent large fluctuations in concentration of unbound drug in the body fluids (d) protein binding reduces diffusion of drug into the cell and there by delays its metabolic degradation e.g. high protein bound drug like phenylbutazone is long acting. Low protein bound drug like thiopental sodium is short acting.
2. Plasma concentration of drug (PC):
It represents the drug that is bound to the plasma proteins (albumins and globulins) and the drug in free form. It is the free form of drug that is distributed to the tissues and fluids and takes part in producing pharmacological effects.
The concentration of free drug in plasma does not always remain in the same level e.g.
i) After I.V. administration plasma concentration falls sharply
ii) After oral administration plasma concentration rises and falls gradually.
iii) After sublingual administration plasma concentration rise sharply and falls gradually.
3. Clearance:
Volume of plasma cleared off the drug by metabolism and excretion per unit time.
Protein binding reduces the amount of drug available for filtration at the glomeruli and hence delays the excretion, thus the protein binding reduces the clearance.
4. Physiological barriers to distribution: There are some specialized barriers in the body due to which the drug will not be distributed uniformly in all the tissues. These barriers are:a) Blood brain barrier (BBB) Blood-brain barrier: To enter the brain, drugs must pass through the endothelial cells of the capillaries of the CNS or be actively transported. By contrast, lipid-soluble drugs readily penetrate into the CNS because they can dissolve in the membrane of the endothelial cells. Ionized or polar drugs generally fail to enter the CNS because they are unable to pass the tight junctions of the endothelial cells = blood-brain barrier (BBB).b) Placental barrier: which allows non-ionized drugs with high lipid/water partitioncoefficient by a process of simple diffusion to the foetus e.g. alcohol, morphine. 5. Affinity of drugs to certain organs: The concentration of a drug in certain tissues after a single dose may persist even when its plasma concentration is reduced to low. Thus the hepatic concentration of mepacrine is more than 200 times that of plasma level. Their concentration may reach a very high level on chronic administration. Iodine is similarly concentrated in the thyroid tissue.
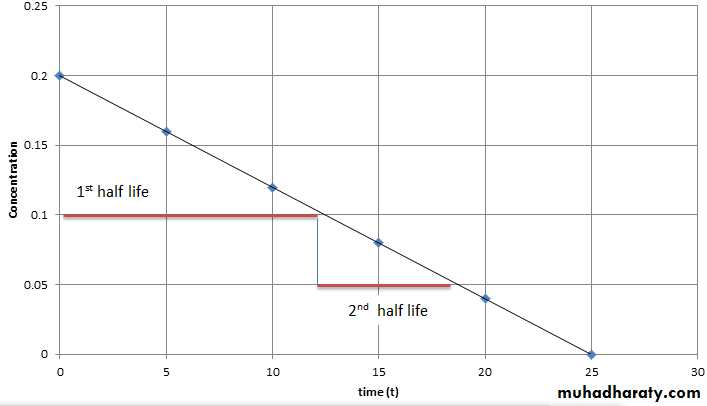
Half-Life (t1/2) defined as the time it takes for blood level of a drug to fall to one-half (50%) of the level measured at some prior time Half life of a drug is directly proportional to the volume of the distribution and inversely proportional to the clearance. Half life = 0.693 x Vd/ total body clearance
Steady State Concentration A steady-state plasma concentration of a drug occurs when the rate of drug elimination is equal to the rate of administration. Any change in drug dose and interval will change the steady state level the rate of drug elimination= the rate of administration
Drug Metabolism (Biotransformation): Sites:Liver is the main organ. Others include GIT, lungs, kidneys, skin, adrenals and blood (plasma).Types of Biotransformation:1. Enzymatic : a. Microsomal and b. Non-microsomal 2. Non-enzymaticNon Enzymatic Elimination:Spontaneous, non-catalyzed and non-enzymatic type of biotransformation for highly active, unstable compounds taking place at physiological pH. Very few drugs undergo non-enzymatic elimination.
Enzymatic Elimination:Biotransformation taking place due to different enzymes present in the body/cells is known as enzymatic elimination. Non-Microsomal Biotransformation:The type of biotransformation in which the enzymes taking part are soluble and present within the mitochondria. eg: Xanthine oxidase converting hypoxanthine into xanthine. Monoamine oxidase involved in non-microsomal metabolism of catecholamines and noradrenaline. Alcohol dehydrogenase responsible for metabolism of ethanol into acetaldehyde Microsomal Biotransformation:Enzymes responsible are present within the lipophilic membranes of endoplasmic reticulum. Enzymes isolated from ER possess enzymatic activity termed as microsomal mixed function oxidase system.Components:Cytochrome P450 (ferric, ferrous forms)NADPH (flavoprotein)
Biochemical Reactions:Phase I reactionsPhase II reactionsPhase I reactions: modificationPhase I reactions are non-synthetic chemical reactions occurring mainly within the ER. The parent drug is converted into more soluble agents by introduction or unmasking of functional component.Phase I reactions include: -Oxidation(Hydroxylation, Dealkylation, O-Dealkylation, N-oxidation, Sulfoxidation, Deamination, Desulfuration)- Reduction ( eg: Chloramphenicol).- Hydrolysis ( eg: Esters: and Amides). Consequences of Phase I reactions:1. Active drug may be converted into inactive metabolite ( mainly). 2. Active drug may be converted into more active metabolite. Eg: morphine is converted into more active metabolite.3. Prodrug may be converted into active metabolite4. Active drug may be converted into toxic metabolite e.g. halothane used in general anesthesia, is converted into trifluoroacetylated compound or trifluoroacetic acid, leading to hepatic toxicity.
Phase II reactions ( conjugation Reaction):Phase II is a conjugation reaction followed the phase I reaction, the products of phase I were conjugated with endogenous substrates like glucuronic acid, sulphuric acid, amino acid or acetic acid, yielding more excretable drug conjugates which are excreted by the kidneys. Phase II reactions lead to :-Usually inactivation of drug- Production of water soluble metabolites, which is the main aim of biotransformation.- Usually detoxification reaction , with some exception ( producing of toxic conjugates eg: methanol is converted into formaldehyde, which is toxic)
Factors Affecting BiotransformationBiotransformation is significantly affected by a number of factors, these include:1. Enzyme Induction:When drugs given over prolonged period of time, upregulation of enzymes takes place. The rate of metabolism increases as enzyme induction takes place. The drugs which bring about these changes are known as enzyme inducers. Some examples include anticonvulsants like phenytoin, carbomycin, chronic alcoholism. various sedatives, hypnotics and tranquilizers. Consequencs of Microsomal Enzyme InductionDecreased intensity and duration of action of drugs e.g. failure of contraceptivesIncreased intensity of action of drugs activated by metabolism. E.g. acute paracetamol toxicity is due to one of its metabolites.If drug induces its own metabolism e.g. cicobarbitone it develops tolerance so effects are not produced.Precipitation of acute intermittent porphyria. Enzyme induction might increase porphyrin synthesis.Intermittent use of an inducer might interfere adjustment of dose of another drug e.g. oral anti coagulants, oral hypoglycemic, antiepileptics and antihypertensives.Auto induction: The phenomenon in which a drug induces metabolism of other drugs as well as its own. eg. carbamazepine-antiepileptic.
2. Enzyme InhibitionThe process in which drug metabolizing capacity of cytochrome is decreased is known as enzyme inhibition. The rate of metabolism is decreased. Drugs bringing about these changes are known as enzyme inhibitors. Examples include ketoconazole- antifungal drug, cimetidine and verapamil- calcium channel blocker.3. Presystemic Metabolism/First pass effect/Route of AdministrationDrugs following first pass metabolism have decreased bioavailability. Most of the drugs are metabolized within the liver. Changing the route of administration might change the first pass metabolism.Propanolol is 80% metabolized before reaching systemic circulation.4. Genetic Variations:Inter individual variations might occur, as drugs behave differently in different individuals due to genetic variations resulting in absent or malformed enzymes due to absent or malformed genes. eg: fast acetylators and slow acetylatorst. 5. Environmental factors:Cigarette smokers, Chronic alcoholism and pesticides might lead to enzyme In hot and humid climate biotransformation is decreased and vice versa. At high altitude, decreased biotransformation occurs due to decreased oxygen leading to decreased oxidation of drugs.
6. AgeExtreme age groups (infants and geriatrics) associated with slow metabolic process. Chloramphenicol (antimicrobial drug) when administered in infant, does not have great efficacy. Toxic effects in the form of grey baby syndrome might occur. The baby may be cyanosed, hypothermic, flaccid and grey in color. Shock and even death might occur if toxic levels get accumulated.Diazepam (sedative hypnotic) may result in floppy baby syndrome in which flaccidity of the baby is seen.In elderly, most metabolic processes were slow down because of decreased liver functions and decreased blood flow through the liver. The drug doses should be decreased in the elderly.7. SexMale have a higher BMR as compared to the females, thus can metabolize drugs more efficiently, e.g. salicylates. Females, during pregnancy, have an increased rate of metabolism. Thus, the drug dose has to be increased. After the pregnancy is over, the dosage is decreased back to normal levels. Example includes phenytoin, whose dose has to be increased during pregnancy (specially second and third trimester).8. Drug-Drug Interaction9. Nutrition 10. Pathological Conditions ( hepatic , cardiovascular, hypothyroidism)11. Circadian rhythm
Elimination of drug is the sum of the processes of removing an administered drug from the body. Elimination may be by:BiotransformationExcretion
Excretion Excretion is the process of removing a drug and its metabolites from the body. This usually happens in the kidneys via urine produced in them. Other possible routes include bile, saliva, sweat, tears and faeces. Most drugs are insufficiently polar (and, therefore, water soluble) to be excreted directly. Instead they need to metabolise to produce more polar, water-soluble molecules. Excretory system The excretory system is made up from the two kidneys, ureters, bladder and urethra, together with the branches of the two renal arteries and veins. Blood passes into the kidney’s nephron (kidney tubule) where three processes can happen: Glomerular filtration: small drug and metabolite molecules and those not bound to plasma protein are filtered from the blood. Large molecules or those bound to plasma protein are poorly excreted by glomerular filtration. Tubular secretion: most drugs enter the kidney tubule by tubule secretion rather than glomerular filtration. The process involves active transport against a concentration gradient and, therefore, requires energy and carriers to transport basic drugs such as dopamine and histamine, and carriers for acidic drugs such as frusemide and penicillin. Tubule reabsorption: Some drugs and metabolites are absorbed back into the bloodstream.This does not require energy. It is passive transport.
Elimination by LiverLiver is the major site for metabolism. It converts lipophilic compounds into hydrophilic compounds by phase I and II reactions, which makes the drugs more excretable. In membranes of canaliculi, transporters for active secretion of drugs or metabolites are present as well. Elimination by LungsLungs constitute the most different route of drug elimination. This is the only route by which lipophilic drugs are excreted because they are absorbed through the alveolar membrane. Examples include general anesthetics, which are gases pumped through the endotracheal tubes and diffuse across the alveolar membrane. When stop their administration, pure oxygen is supplied. The body acts as a reservoir and transport occurs in reverse. Thus lipophilic compounds are lost through the lungs. Alcohol breadth is another example which can be tested by alcohol breath test, by which alcohol in the excreted air is measured.IntestinesDrugs are mostly absorbed in the small intestine. Anthracene purgatives, which act mainly on the large bowel, are partly excreted in to that area from the blood stream after absorption from small intestine. Heavy metals are also excreted through the intestine and can produce intestinal ulcerations.
Minor SourcesBreast milk is important because many drugs are excreted in it. Some effects of the drugs may be transferred to the baby, which may prove harmful. It is important to know which drugs are not to be used during breast feeding. Milk being slightly acidic than plasma, weak bases get ionized and have equal or higher concentration in milk than in plasma. Non electrolytes like ethanol and urea readily enter the milk independent of pH. 70% of plasma concentration of tetracyclines may enter milk, prolonged usage of which might cause permanent staining of teeth and weak bones in the baby. Ampicillin may lead to diarrhea and allergic sensitizations. Chloramphenicol might lead to aplastic anemia in baby, bone marrow suppression and grey syndrome. Morphine, opoids and smoking may cause lethargic baby.If the mother is taking drugs, she should lactate the baby a few hours after taking drugs or most preferably half an hour before intake. Sweat glands.
Clearance of drugUnit volume of blood which is cleared off a drug per unit time is known as clearance.Clearance is not a measure of how much drug is being eliminated; it is only a measure of how much plasma is cleared of it per minute. Units are ml/min, sometimes ml/min/kg body weight are used. Rate of elimination = Quantity or volume of urine measured (ml/min) X conc. substances in urine (mg/ml)Rate of elimination = ml/min x mg/ml = mg/min Clearance= rate of elimination of substance in urine/ concentration of the drug in the blood.Clearance = mg/min / mg/ml = ml/minTotal clearance = CL kidney + CL liver + CL…..