GENETICS
Heredity is the transmission of information required to construct multiple proteins. These proteins have diverse roles and different subsets, they are utilized by different cell types but all are encoded in the cells DNA which is organized into discrete structures called chromosomes.Chromosomes: every cell nucleus contains a set of chromosomes. Each chromosome consists of a single molecule of DNA (deoxyribonucleic acid) together with associated acidic and basic proteins.
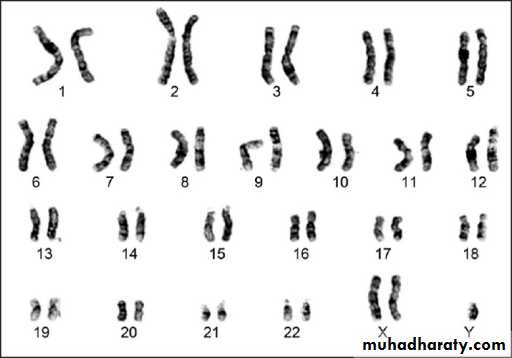
Most human cells contain 46 chromosomes (the diploid number) with 22 pairs of autosomes which are a like in males and females and a pair of sex chromosomes: XX in female and XY in a male.
Each chromosome has a narrow waist called the centromere which has a constant position for a given chromosome. The centromere divides each chromosome into short and long arms.
At mitosis each chromosome replicates to form a pair of sister chromatids which are held together at the centromere. Although exchanges of genetic material can occur by crossing over (sister chromatid exchange) during mitosis, as each sister chromatid is identical, clinical consequences do not arise. Thus at the end of cell division each daughter cell has identical set of 46 chromosomes.
In contrast, reduction cell division or meiosis results in cell with a half set (haploid number) of 23 chromosomes. Meiosis, which is confined to gonadal cells involved in gametogenesis, consist of two successive division in which the DNA replicates only once before the first division. Each mature egg thus normally contains one of each pair of autosomes and one X and each mature sperm has one of each pair of autosomes and the X or Y chromosomes. At fertilization the diploid number is restored and in consequence half of each individual's autosomes are derived from each parent and a female has an X from each parent. Whereas a male has a maternal X and a paternal Y sex chromosome.
DNA: each molecule of DNA is composed of two nucleotide chains which are coiled clockwise around one another to form double helix. Each nucleotide consists of a nitrogenous base, a molecule of deoxyribose and a phosphate molecule.
The nitrogenous bases are of two types, purines and pyrimidines. In DNA there are two purine bases, adenine (A) and guanine (G) and two pyrimidine bases, thymine (T) and cytosine (C). The nucleotide chains run in opposite direction and are held together by hydrogen bonds between A and T or between G and C since A: T and G: C pairing is obligatory the parallel strands must be complementary to one another.
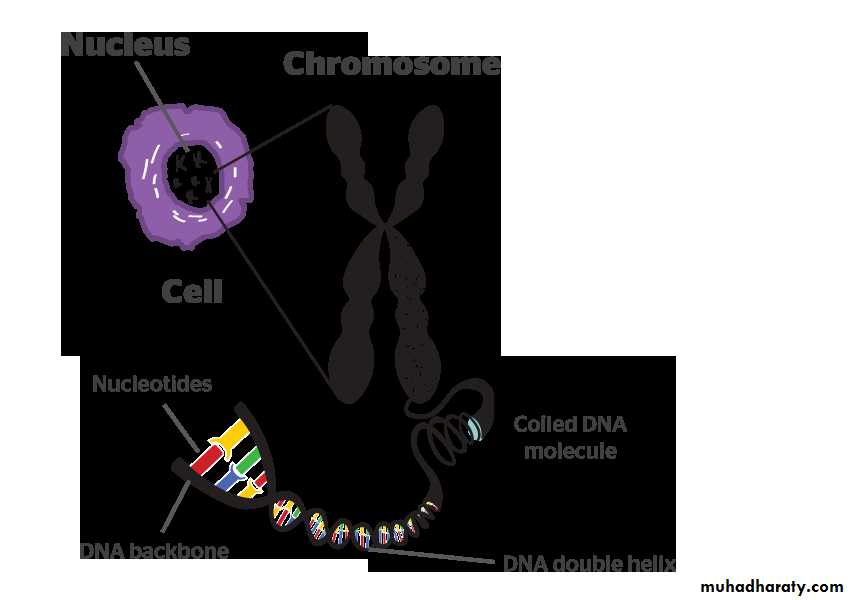
Gene: is the unit of the DNA which codes for a protein.
Genetic diseases:-Are large group of diseases can be subdivided into:-
Single gene defect (dominant & recessive).
Chromosomal diseases (numerical & structural)
Multifactorial diseases & non traditional disorders.
Mutation refers to permanent changes in the DNA. Those that affect germ cells are transmitted to the progeny and may give rise to inherited diseases. Mutations in somatic cells are not transmitted to the progeny but are important in the causation of cancers and some congenital malformations
Thus there is certain changes occur in nitrogenous bases may lead to abnormal protein synthesis.
Single gene diseases (Unifactorial diseases):-These are caused by a mutation in a gene. Genes may behave as dominant, i.e. when one of the alleles becomes mutated it results in a genetic disease; or they may behave as recessive, i.e. the diseases does not manifest unless both alleles are affected by the same mutation.
A third category of genes are those which determine an autosomal character but are situated on the sex chromosome (sex-linked)
The question arises why some genes act in a dominant manner while others behave in a recessive fashion, i.e. the problem of dominance and recessiveness. To answer this question, one has to consider and always remember the following principles:
A Single gene is responsible for formation of a single type of protein, but since proteins are made of units of polypeptides that could be the same or different in one molecule of protein, the principle becomes:
A Single gene is responsible for the formation of a single type of polypeptides, and if we know that our body structures and functions from the moment of post-fertilization to the full maturity and later on are determined by proteins one can understand how genes function. These types of proteins are varied; they could be
Structural proteins, like fibrous tissue and elastic tissue proteins
Immunoglobulins
Signal proteins produced by many oncogenes
Receptors
Enzymes
Hormones
Therefore, the action of the gene being dominant or recessive is determined by the type of protein it produces and its function.
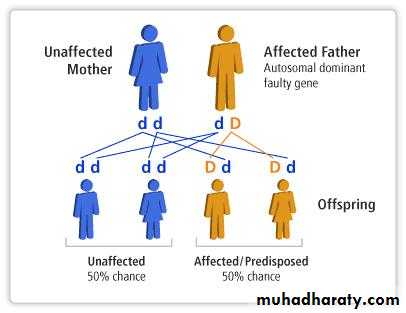
Autosomal dominant disorders:
are manifested in a heterozygous state (when one of the alleles becomes mutated)one of the parents of the affected individual should be affected and the child appears as diseased individual.
both males and females are affected and both can transmitted the condition. When affected person marries unaffected one, every child has one chance in two of having the disease.
Some times the parents are normal but the child is diseased that happened because the mutation occur in the cell of that child alone while then parents are completely normal. The siblings of this child are neither affected nor at risk of developing the disease
Some trait is seen in all individuals that carrying the mutant gene but it is expressed differently among different individuals: phenomena called variable expressivity.e.g. polydectaly may be expressed in toes or in fingers as one or more digitis.
Dominant genes usually produce two types of proteins:
Major structural proteins, which form or are present in many parts of the body e.g.is Marfan syndrome in which there is mutation in fibrillin gene leading to a qualitative & quantitative defects in fibrillin which result in skeletal abnormality .
Enzymes, which are key enzymes in metabolic pathways, under feedback mechanism, or receptors regulating metabolic pathways.
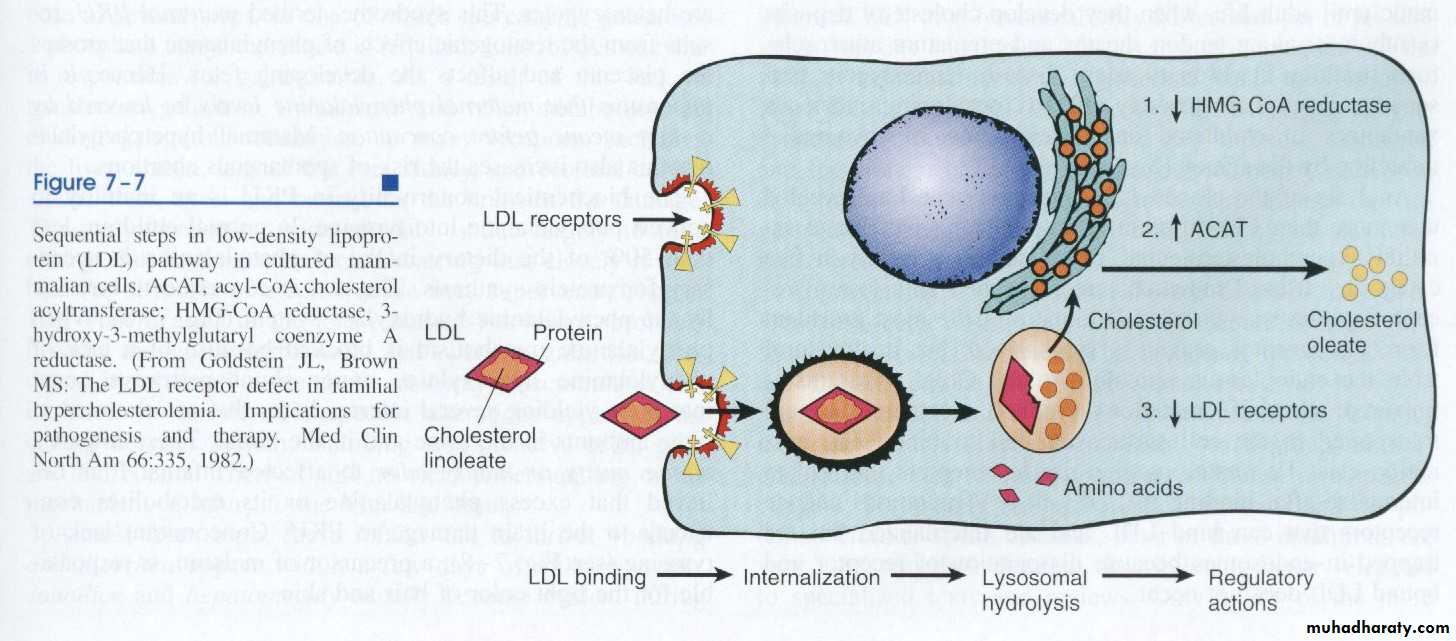
Example of the second is AD familial hypercholesterolemia [] disease where the receptors for LDL are mutated. They are responsible for regulation of LDL in the cells and the circulation. To explain the latter example and how the action of this pathway is executed, let us consider the pathway of circulating LDL: usually it should enter the cells of the body for building cellular membranes and nuclear membranes to replace the old ones that are affected by wear and tear. LDL could not enter the cells unless it is complexed with receptors on the cell membrane. Once it is inside the cell, the complex will be degraded into free cholesterol and amino acid. The latter is the remnant of the proteinaceous coat of the lipoprotein. The free cholesterol in the cell constitutes the cholesterol pool of the cell and its level is regulated by three systems of enzymes, the HMG-CoA reductase, which forms cholesterol from fatty acids, ACAT, which hydrolyzes cholesterol into esters and thus rendering it inactive and the number of the receptors on the surface of the cell. If the pool concentration is low, messages are sent to activate the HMG-CoA, to inactivate ACAT and increase the number of receptors on the cell surface. Therefore, when one of the two alleles responsible for the formation of the receptor protein becomes mutated, half of the number of receptors are formed only, so 50% of LDL which is used to be internalized inside the cell will remain in the circulation unable to enter the cells and a state of hypercholesterolemia results with reading of 400-500 iu/dl of cholesterol in the blood (normal value 180-220 iu/dl)
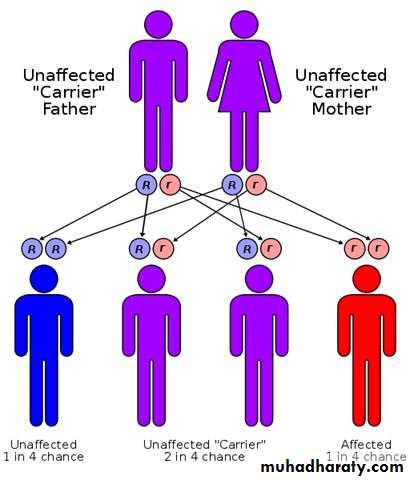
Autosomal recessive disorders:
are manifested in a homozygous state (they occur when both of the alleles at a given gene locus are mutants).usually the parents are unaffected clinically because each has only one mutant gene and so they are a carrier or heterozygote.
For two carrier parents the chance for getting an affected child is 1 to 4.
As for recessive genes, they are protein enzymes, which usually share in catabolic pathways and when both alleles are ?defective, there is no protein, i.e. no enzyme and therefore the catabolic pathway is obstructed with the accumulation of the biochemical substrate. Examples of these are mucopolysaccharidosis & phenylketonuria (PKU) and most of inborn errors of metabolism.
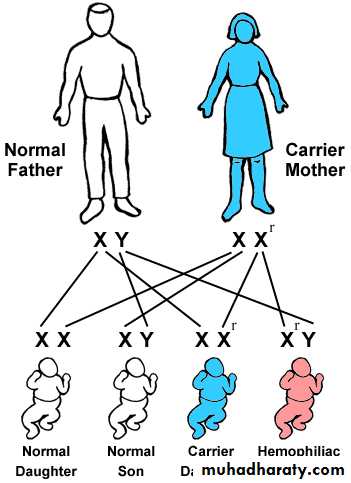
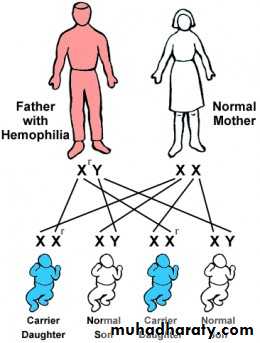
Athird category of single gene disorders are Sex-linked diseases in which genes that determine an autosomal character are situated on the sex chromosome (sex-linked) and because most of the genes are carried on the “X” and very few are present on the “Y”, usually sex-linked is used for “X”-linked both dominant and recessive. Most of the X-linked disorders are X-linked recessive and are characterized by the following features:
They are transmited by heterozygous female carriers only to sons.
An affected male does not transmitted the disorder to sons but all daughters are carriers.
Sons of heterozygous women have one chance in two of receiving the mutant gene.
Sex linked dominant disease e.g. is the vit D resistant rickets
Y inheritance disease is the hairy ears in males
Sex-linked diseases, because most of the genes are carried on the “X” and very few are present on the “Y”, usually sex-linked is used for “X”-linked both dominant and recessive. In this type of inheritance, there is a lot of deviation from the expected and their explanation is forwarded by a hypothesis known as Lyon’s hypothesis, e.g. in clinical practice, both haemophilia and G6PD-deficiencies are diseases caused by sex linked genes recessive in nature, i.e. only males who carry the mutated gene on their “X” are affected clinically while carrier females are usually silent clinically but transfer the disease to their sons. But it happens that some cases of both diseases present in female by Lyon’s hypothesis, which states that in a female’s autosomal cells, all the “X” chromosomes will be inactivated except one which remains active during inetrphase.
This process of inactivation takes place early n the post-fertilization period, 19-20 days P.F.
The process of inactivation is random concerning the origin of the “X” inactivated, i.e. paternal “X”, which comes from the father or maternal; “X” that comes from the mother.
In a cell, all the daughter cells that descend from it, the same “X” will remain inactive.
This means that 50% of the “X” chromosomes are inactivated but this does not necessarily involve all the paternal “X” or all the maternal “X”. in some areas the paternal X is being inactivated while in other areas, it is the maternal “X” that are being inactivated, therefore, the body of the female is a mosaic concerning the function of the active “X”. So, a heterozygote female for type G6PD enzyme A & B; if we examine different parts of her body for the type of the enzyme, we either find type A or type B and never both in one part of the body. In contrast males could either be A or B.
A female who carries the mutated gene for “X” linked and presents clinically the disease; it happens by chance that in most parts of her body the “X” that carries the mutated gene remains active, which results in deficiency of the product of the gene disease. This is because of the randomness of the inactivation. Those females are known as manifesting carriers in clinical practice.
Etiology:
Mutation refers to permanent changes in the DNA. Those that affect germ cells are transmitted to the progeny and may give rise to inherited diseases. Mutations in somatic cells are not transmitted to the progeny but are important in the causation of cancers and some congenital malformations
Thus there is certain changes occur in nitrogenous bases may lead to abnormal protein synthesis.
All single gene diseases are due to mutations. They are of different types:
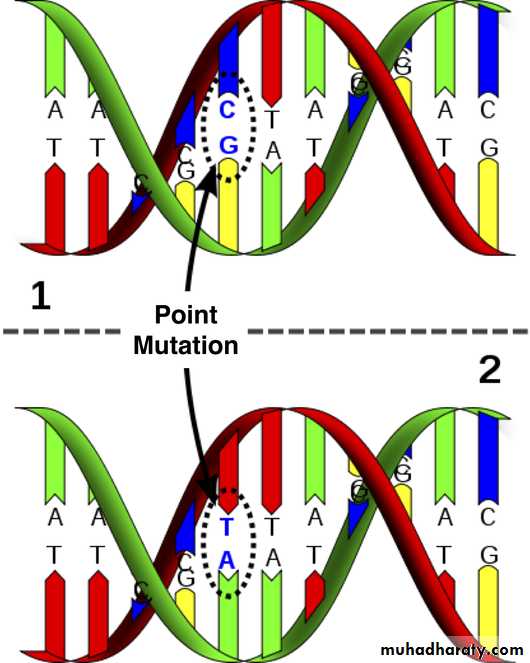
Single point mutation which is the commonest. They usually result from a change in one of the nucleotide bases that form the trios (three bases), each of which codes for a specific amino acid in the protein molecule. Not all of these changes will result in a mutation that causes a disease.
2-Additional – deletional mutations : They could be one of three types :
Addition or deletion of a single base.Addition or deletion of 3 bases or the multiple of 3,
Addition or deletion of a large piece of DNA inside the gene (intragenic) or in between the genes (intergenic). Again this creates variability and it is used for genetic testing and diagnosis of genetic diseases.

Chromosomal Diseases
These are classified into:a. Numerical abnormalities, which is defined as a gain or loss of a whole chromosome from the usual number of chromosomes in the karyotype (i.e. 46 chromosomes). Gain or loss in the sex chromosomes especially the X-chromosome is compatible with life and is relatively common; while loss of an autosomal chromosome is usually non-viable and a fertilized ovum carrying such karyotype could not sustain pregnancy to full term and usually are lost very early in pregnancy, i.e. abortion.
Autosomal chromosome trisomy is exemplified by trisomy 21 or Down’s syndrome, trisomy 18 or Edward’s syndrome, trisomy 13 or Patau’s syndrome.
Sex chromosome trisomy is exemplified by Klienfilter’s syndrome in male and triple X (XXX) syndrome in female or XYY syndrome in male, while monosomy of sex chromosome is when a female loses one X resulting in Turner’s syndrome, 45 X known as aneuploidy.
b. Structural abnormalities: it usually results from breakage followed by rearrangement of material (a cell suffering from structural abnormality has the normal number of 46 but the chromosomes are morphologically or structurally abnormal).
These abnormalities are of different types:
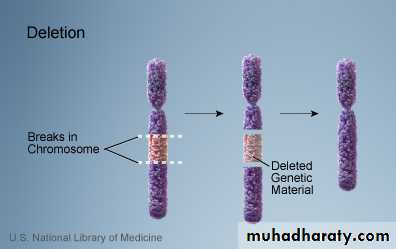
A. Deletion: loss of a piece of a chromosome. It is of two types
i. Terminal: single break may delete a terminal segment.
ii. interstitial: where the piece of a chromosome between two breaks is lost resulting in a syndrome
B. Inversion:
This abnormality results from two breaks through out the length of the chromosome which either involve the centromere area or not and the piece between the two breaks will rotate 180o before it returns to its place. So the genetic piece which is broken it will rotated so the position of the gene occupied by this segment is abnormal & the content of the genetic material are changed according to the piece which is inverted & this will result in abnormal fetus with signs & symptoms of abnormality.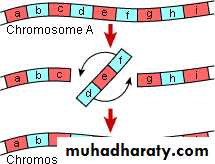
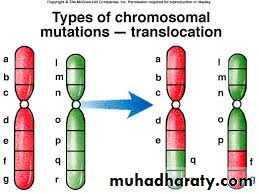
C.Translocation: This is defined as exchange of segments of chromosomes between two non-homologous chromosomes so there is a translocation of some oncogenes from their normal habitat to a new situation where they are induced to function in uncontrolled manner leading to malignancies. This is usually seen in leukemias and lymphomas, e.g. Philadelphia chromosome.
D. Isochromosome: This abnormality results from aberrant division of the centromere which is the last part of the chromosome that divides in the mitosis to separate the two sister chromatids into individual chromosomes. This aberrant division takes place in a horizontal way rather than the perpendicular natural way. So the resulting two chromosomes are imbalanced, one formed of two short arms and the other of two long arms. Each of them is an isochromsome.

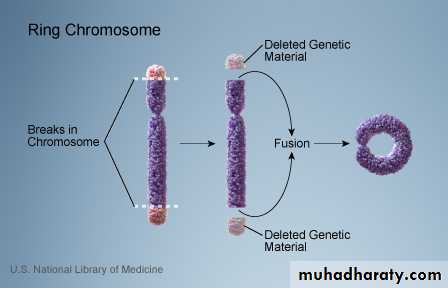
E. Ring-chromosome: It results from deletion of both ends of a chromosome and then the ends, because of the adhesive nature of the exposed DNA, will stick together forming a ring.
Cytogenetic disorders involving autosomes:
Down syndrome (Trisomy 21), karytype (47, xx or xy, +21):is the most common of the chromosomal disorders. About 95% of affected persons have trisomy 21 resulting from meiotic nondisjunction. The parents of such children have normal karyotype and are normal in all respects. Increasing of maternal age has a strong influence on the incidence of Down syndrome. The correlation with maternal age suggests that in most cases the meiotic nondisjunction of chromosome 21 occurs in the ovum.
In about 4% of all patients with trisomy 21, the extra chromosomal material is due to translocation of the long arm chromosome 21 to chromosome 22 or 14 and the remaining 1% of trisomy 21 patients are mosaics.
Trisomy 21 is the leading cause of mental retardation. The patient has a combination of epicanthic folds and flat facial profile which is quite characteristic. Congenital malformations are common. Approximately 40% of patients have cardiac malformations, which are responsible for most of the death in early childhood. They found that approximately 80% of those without congenital heart disease can expect to survive 30 years but most of them develop Alzheimer disease and frank dementia.
Serious infections and increased risk of developing acute leukemias are another cause of morbidity and mortality in patients with trisomy 21.

Cytogenetic disorders involving sex chromosome:
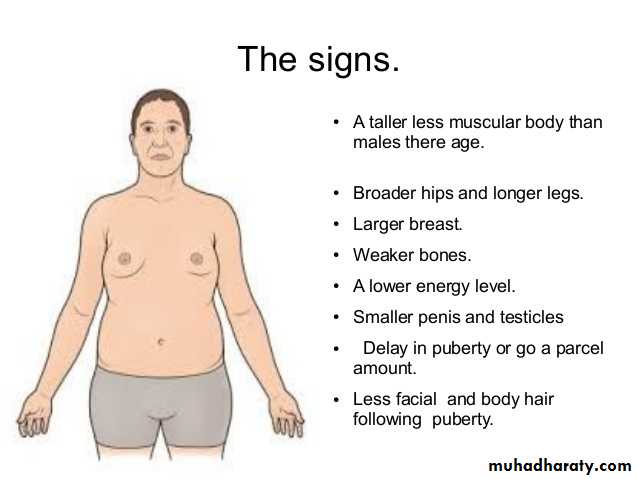
Klinefelter syndrome: this syndrome is best defined as male hypogonadism that develop when there are at least two X chromosomes and one or more Y chromosomes. Most patients are 47, XXY. This karyotype results from nondisjunction of sex chromosomes during meiosis. The extra X chromosome may be of maternal or paternal origin. Advanced maternal age and history of irradiation of either parent may contribute to the meiotic error resulting in this condition. Approximately 15% of patients show mosaic patterns (i.e. 46,XY/47,XXY or 47,XXY/48,XXXY) and the presence of a 46,XY line in mosaics is usually associated with a milder clinical condition.Although the following description applies to most patients, it should be noted that klinefelter syndrome is associated with a wide rang of clinical manifestations. In some it may be expressed only as hypogonadism, but most patients have an increased length between the soles and the pubic bone, which creates the appearance of an elongated body. Reduced facial body and pubic hair with gyneocomastia are also frequently noted. Testicular atrophy, the serum testosterone levels are lower than normal while urinary gonadotropin levels are elevated. So the principle clinical effect of this syndrome is sterility. Only rarely the patients are fertile and these are presumably mosaics with a large proportion of 46, XY cells. This syndrome may be associated with mental retardation but the degree of intellectual impairment is typically mild and in some cases is undetectable. The reduction in intelligence is correlated with the number of extra X chromosomes. Thus, in patients with the most common variant (XXY), intelligence is nearly normal, but in those with rare variant forms involving additional X chromosomes, significantly subnormal levels of intelligence, as well as more sever physical abnormalities are found.
Turner syndrome:
It is characterized by primary hypogonadism in phenotypic females, results from partial or complete monosomy of the short arm of the X chromosome. In approximately 57% of patients, the entire X chromosome is missing, resulting in a 45,X karyotype. These patients are the most severely affected. Typical clinical features associated with 45X Turner syndrome include significant growth retardation, leading to abnormal short stature; swelling of the nap of the neck due to distended lymphatic channels (in infancy) that is seen as webbing of the neck in older children; low posterior hair line; shieldlike chest with widely spaced nipples; high arched palate; lymphedema of the hands and feet; and a variety of congenital malformations such as horseshoe kidney, bicuspid aortic valve and coarctation of the aorta. Affected females develop normal secondary sex characteristics; the genitalia remains infantile, breast development is minimal and little pubic hair appears. Most have primary amenorrhea, and morphologic examination reveals transformation of the ovaries into white streaks of fibrous stroma devoid of follicles. The mental status of these patients is usually normal. Curiously, hypothyroidism caused by autoantibodies is noted in 25% to 30%. In adult patients a combination of short stature and primary amenorrhea should prompt strong suspicion of Turner syndrome. The diagnosis is established by karyotyping.Approximately 43% of patients with turner syndrome either are mosaics (one of the cell lines being 45,X) or have structural abnormalities of the X chromosome.
Congenital malformations:
This topic is discussed within the context of genetic diseases because it has some bearings to those diseases. Not all congenital malformations, as it may come to the mind of the student, are caused by genetic aetiology; but only a small percent of them are so. The majorities of congenital malformations are caused by environmental aetiology, and from this angle comes their relationship to genetic diseases.
If congenital malformations are represented by a big circle (that encompasses genetic disease in addition to others) Figure (1), the largest group i.e. 2/3 of the surface area is of unknown aetiology, while only a small sector represents diseases of genetic basis.
Definition:
It is a deformation of structure or function of an organ present at birth.
It may be on the surface of the body, e.g. cleft lip or inside the body, e.g. horseshoe kidney; it may be macrocellular, i.e. recognized by the naked eye e.g. club foot or microcellular i.e. recognized only by microscopical examination, e.g. sponge kidney where the defect is abnormal connection between the collecting tubules and the urineferous tubules leading to thin microscopical dilatation that gives the kidney a spongy feeling; it could be diagnosed by mere naked eye, e.g. microcephaly [] or microphthalmia [] or it may need special procedure for diagnosis, e.g. congenital heart defects; it may present at birth showing signs and symptoms, e.g. tracheo-esophageal fistula or the signs and symptoms may present later in life (but the defect is actually present at birth), e.g. adult polycystic kidney; it may be familial or non-familial, i.e. either there are multiple cases in one family or it is the only case in the family (sporadic); and lastly it could be genetic, i.e. direct descent from parents through genetic defect or could be non-genetic, i.e. caused by environmental causes, e.g. microcephaly; some cases are genetic while others are due to uterine infection during pregnancy (prenatal infection).
Causes
The etiological factor that results in congenital malformation is known as a teratogen. Some teratogens may act as mutagen or carcinogen.
The effect of a teratogen is most effective on growing tissue with rapid division rate but not all teratogens affecting different foeti cause the same severity of action, i.e. the action of one teratogen is variable in different foeti. This is due to the following factors
Dose of the teratogen: larger doses of course causes severer effects.
Time of exposure to the teratogen, e.g. the action of the Mullerian inhibiting factor (MIF) which is an enzyme produced by the foetal testis at a certain time (7th week post-fertilization) to effect the regression of the female primitive reproductive organ in a male foetus. If the testis fails to produce this enzyme at that time, then it will have no effect and the female reproductive organs will remain inside the abdomen of the male infant resulting in ambiguous genitalia.
Host susceptibility: some foeti are more susceptible than others for the same dose and therefore they are more severely affected.
Interaction with other factors, environmental or genetic constitution of the foetus.
Types of teratogens
I. Genetic, these could be:
1. Single gene that is either:
a. autosomal dominant (AD).
b. autosomal recessive (AR).
c. sex-linked dominant (XD).
d. sex-linked recessive (XR).
2. Chromosomal abnormality that could be:
a. numerical.
b. structural.
II. Environmental teratogens; these causes:
In-utero infection :
Prenatal infection of the fetus with bacteria, viruses, or parasites may occur during pregnancy due to maternal infection that is transferred to the fetus through the placenta causing fetal infection. The most common is the viral infection with rubella virus (German measles), and influenza virus.
Rubella usually causes severe malformation the earlier it infects the fetus The modern immunization against rubella of girls at reproductive age is one of the very successful methods to prevent this type of malformation.
Usually, rubella causes congenital heart defect, mainly septal defects, microcephaly, cataract [], or infection of the chambers of the eyes leading to blindness, deafness and mental retardation.
Influenza virus usually causes cleft lip and palate [] and neural tube defects
Toxoplasmosis is a parasite infestation contracted from animals, usually sheep or cats; it may cause microcephaly, jaundice, and mental retardation.
Physical teratogens :
These could be in the form of:Heat, whether sauna bathing, or from weather or fever (if it does not cause abortion) usually causes neurological abnormalities and neural tube defects.
Physical pressure that may be caused from inside the uterus or outside it, pressing the growing foetus and thus preventing its proper growth.as uterine fibroid.
Ionizing radiation, whether diagnostic or therapeutic. Radiation or accidents usually causes malformation. Radiation causes microcephaly, anophthalmia and spina bifida. The most sensitive period for the foetus is the 1st two weeks post-fertilization to the 4th-5th weeks of conception.
Chemical teratogens, these could be :
Non-medicinal: like insecticide, and household chemical used in cleaning and industrial chemical. Some of those usually contain organic phosphorous, which is very toxic. These usually are ingested accidentally to contaminate food and water, also alcohol and cigarette smoking in this category, both cause intrauterine growth retardation and also delayed mental development in later life after birth with small stature.
Medicinal chemicals : one should consider that any drug is not safe during the early weeks post-fertilization and medication should be taken cautiously of the most properly documented drug in causing congenital malformation is :
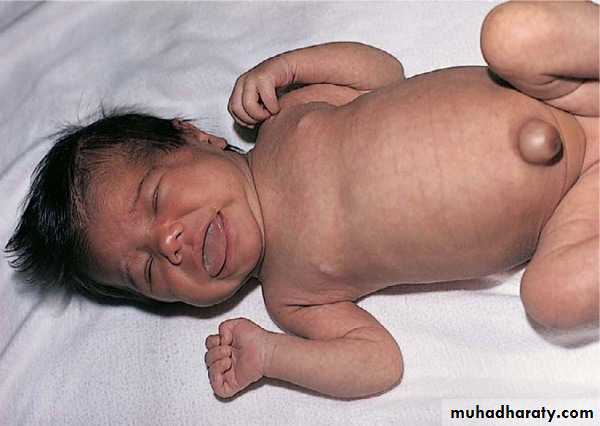
Thalidomide: a drug used for sedation of hyperemesis gravidarum, usually caused amelia and phocomelia
Anti-convulsant, Anticoagulant, Cytotoxic drugs and hormones: like progesterone, estrogen and cortisone.
Maternal disorders
Diabetes, whether treated or not. Cause macrosomia of fetus
Maternal phenylketonuria (PKU), which is a genetic disease that could be treated. Females may reach reproduction with normal state but their high phenylalanine that results from their deficiency of phenylalanine hydroxylase enzyme will destroy the developing brain to cause mental retardation in the fetus.
Thyroid: maternal hypothyroidism will cause a high degree of thyroid stimulating hormone that will suppress the fetal thyroid gland resulting in a state of hypothyroidism after a short period of hyperstimulation (cretinism).