
Immunology / Immunodeficiency
Objectives
The objectives of this lecture are to know
1. Types of Immunodeficiency.
2. T-cell Immunodeficiency.
3. B-cell Immunodeficiency
Immunodeficiency
Definition
Immunodeficiency means failure of immune system to protect the host
from infectious agents or malignant cells or from autoimmune diseases
is known as. It may be confined to one or more than one factor or cell of
the immune system including complete absence or deficiency of that
element. The immunodeficiencies should be suspected in every patient,
who develops recurrent, persistent, severe or opportunistic infections.
Types of immunodeficiency
1. Primary immunodeficiency.
2. Secondary immunodeficiency.
1- Primary immunodeficiency:
Inherited genetic defect in one of the genes responsible for expression
of an immune factor, or impairment of function or absence of immune
cells. Interfere with the innate or the adaptive immunity.
These defects may be:
asymptomatic and discovered accidentally or expressed as severe clinical
conditions at any stage of patient’s life.
lect:4
Dr. Khalid Waleed
M.B.ch.B., Msc., PhD. Immunology

2- Secondary immunodeficiency: It is usually due to impairment of the
immune system by external factor as: Infection
e.g.: acquired
immunodeficiency syndrome (AIDS). Exposure to chemical agents as
chemotherapy (immunosuppressive )
Primary immunodeficiency (PID)
More than 150 different types of primary immunodeficiencies (PIDs) in
the world.
I. Primary T–cells deficiencies:
It usually leads to combined immunodeficiency (CID) in which there is
defect in both T-cell functions and disruption of antibody immune
responses. It includes:
1. Severe combined immunodeficiency (SCID)
2. Thymic hypoplasia [DiGeorge syndrome; 22q11 deletion syndromes]
3. Wiskott-Aldrich Syndrome (WAS)
4. Chronic mucocutaneous candidiasis
5.
Hyper-IgM syndrome
Severe combined immunodeficiency (SCID)
1. This is the most severe immunological defect.
2. It is caused by a large number of rare genetic disorders which lead to
failure of development of the lymphoid system.
3. It usually presents during the first few months of life with failure to
thrive, persistent oral Candida infection, intractable diarrhoea, often
due to persistent viral infection.
4. The humoral immune response defects may appear later than 6 months
age because of the presence of passive maternal antibodies that are
received through the placenta or breast milk.

5. The SCID is classified clinically on the basis of the presence or
absence of B cells and NK cells.
6. The commonest cause of SCID (50%) is due to mutations in the
common gamma chain of the receptors for the cytokines IL-2, IL-4,
IL-7 and others. These diseases are either inherited as autosomal
recessive or they are X-linked diseases.
7. Gene therapy has been used as treament method, where the patient’s
own stem cells are removed from the bone marrow, transfected with
the normal gene using a viral vector and then re-introduced into the
patient.
Thymic hypoplasia [DiGeorge syndrome; 22q11 deletion syndromes]
The thymus, parathyroid glands and parts of the face, jaw and great
blood vessels develop from the third and fourth pharyngeal arches. In the
DiGeorge syndrome their development is arrested, with the result that
affected individuals lack a thymus. Defect is associated with the deletion
of the gene on chromosome 22.
The T-cell deficiency is variable, depending on how badly the
thymus is affected. Affected infants have distinctive facial features; their
eyes are widely separated. They also have congenital malformations of
the heart or aortic arch and neonatal tetany due to hypoplasia or
aplasia of the parathyroid glands. Treatment is by supportive therapy, or
thymic epithelial transplant.
Wiskott- Aldrich Syndrome (WAS) is characterized by:
1. X-linked recessive immunodeficiency disease affects males.
2. Small, abnormal platelets with thrombocytopenia, which may lead to
fatal bleeding.

3. Frequent and severe dermatitis
4. Repeated severe pyogenic and opportunistic infections.
5. Patient’s serum contains high levels of IgA &IgE, normal levels of
IgG and low levels of IgM.
6. Their T cells are defective in function.
7. Splenectomy is useful in reducing the risk of bleeding.
Immunoglobulin replacement therapy is valuable in reducing the risk
of infection.
8. All the features of this syndrome can be corrected by bone marrow
transplantation.
Chronic mucocutaneous candidiasis (CMC)
• Primarily, CMC appears to be associated with defects in IL-17 signaling
• Chronic infection of skin, mouth and nails with Candida.
• Most patients have normal serum immunoglobulins,
• High levels of anti-Candida antibody and impairment of the T-cell
response to Candida.
• Chronic use of anti-fungal therapy is usually required to control the
main manifestations.
Hyper-IgM syndrome
1. This X-linked condition is characterized by a profound lack of IgG
and IgA but an increased level of polyclonal IgM.
2. These patients often present with Pneumocystis jirovecii and other
opportunist infections such as Cryptosporium and are prone to
pyogenic infections.
3. Treated with bone marrow transplantation.

II. B-Cell Immunodeficiencies
Exhibit depressed production of one or more antibody isotypes.
Immunodeficiency disorders caused by B-cell defects ranging from the
complete absence of mature recirculating B cells, plasma cells, and
immunoglobulin, to the selective absence of only certain classes of
immunoglobulins.
X-linked hypogammaglobulinaemia : Bruton’s disease (XLA)
1. The XLA occurs in approximately in 1 200,000 newborns.
2. Occurs due to mutation in Bruton tyrosine kinase, (BTK). The
defect blocks pre-B cell differentiating into B cells and is due to the
lack of a B cell-specific tyrosine kinase, (BTK). The B-cells in bone
marrow arrested at pro-B to pre-B cell stage. B-cells in the circulation
less than 1%.
3. There is a complete absence of serum immunoglobulins.
4. Affected males usually present with recurrent infection at between 6
months and 2 years of age, as they are protected from earlier infection
because of the placental transfer of maternal IgG antibody.
5. Haemophilus
influenzae,
Streptococcus
pneumonia
and
staphylococci are the most frequent causes, and the respiratory tract
and skin the most frequent sites of infection.
6. A combination of immunoglobulin replacement therapy, vigorous use
of antibacterial agents are suggestive measures of treatment.
Common variable immunodeficiency (CVID)
1. There are defects in T-cells signaling to B-cells with impairment of B-
cells response to antigens. The B-cells numbers are normal. Affect
b
oth male and females.

2. It usually presents in later childhood or early adult life and has also
been referred to as late-onset hypogammaglobulinaemia.
3. Characterized by recurrent infection, marked by reduction in the
levels of one or more antibody isotype and impaired B-cell
responses to antigen.
4. There is low IgG and IgA and/or IgM
5. S. pneumoniae, H. influenzae, Mycoplasma spp. are the most common
pathogens.
6. Herpes zoster infection, meningitis, osteomyelitis and skin sepsis also
occur.
7. Immunoglobulin replacement, antibacterial agents and drainage of
infected sites with careful follow-up concerning the other
complications to which these patients are prone are suggestive lines of
treatment.
Selective IgA deficiency:
1. Frequency of disease is 1:700.
2. Unknown genetic cause.
3. IgA producing B-cells cannot differentiate into plasma cells.
4. Typically exhibit normal levels of other antibody isotypes.
5. Clinical picture is variable from asymptomatic in 70% of cases to
severe infections.
6. Allergies and autoimmune diseases.
7. GIT and RT (The primary sites for production of IgA) are most
common sites to get infected.
References:
1. Clinical immunology: principles and practice 5
th
Edition.
2. How the immune system works: 5
th
Edition.
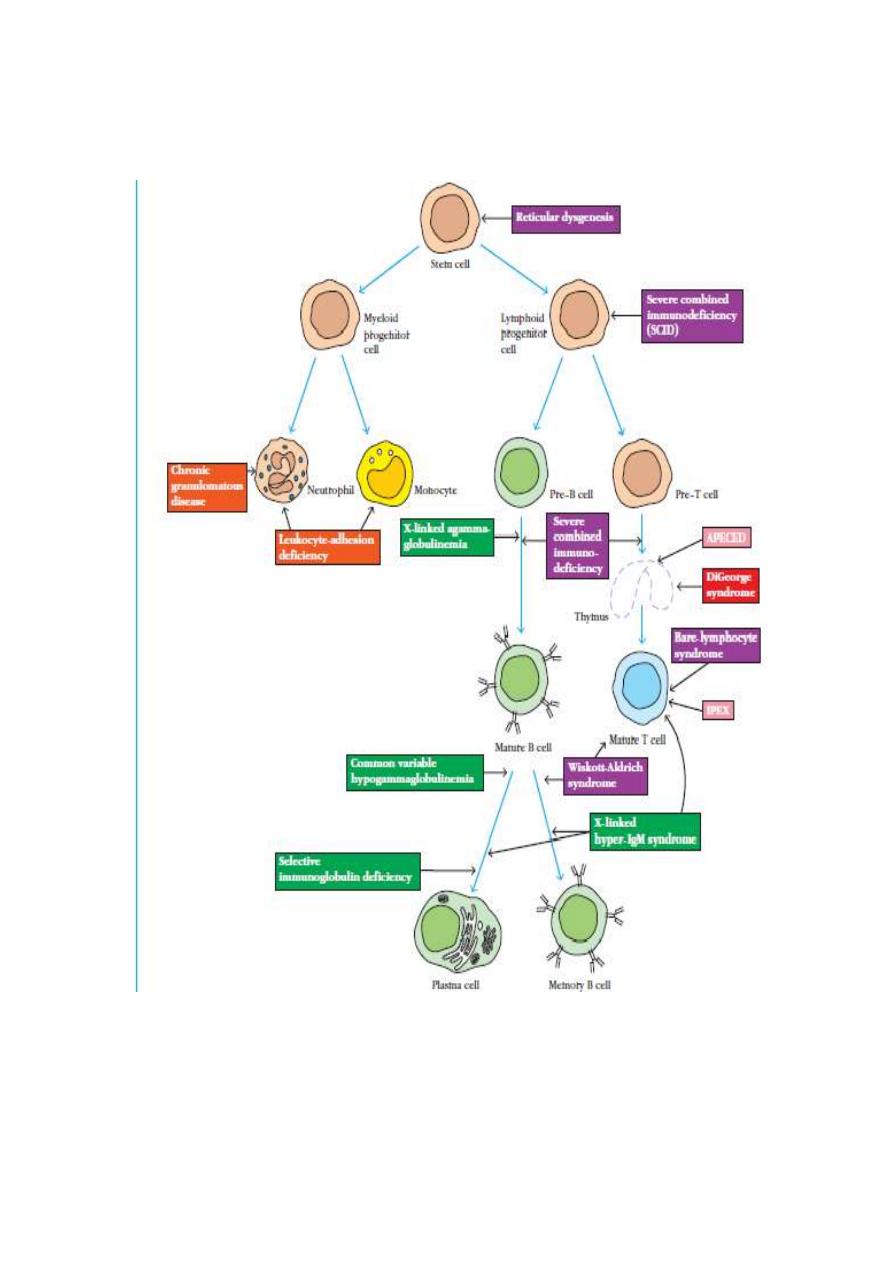
Primary Immunodeficiencies Result from Congenital Defects in
Specific Cell Types
