Pharmacokinetics of Drug Absorption
The pharmacokinetics of drugs following intravenous drug administration are more simple to model compared to extravascular delivery.Extravascular delivery routes, particularly oral dosing, are important and popular means of drug administration. Unlike intravenous administration, in which the drug is injected directly into the plasma, pharmacokinetic models after extravascular drug administration must consider systemic drug absorption from the site of administration, eg, the lung, the gut, etc., into the plasma. Extravascular drug delivery is further complicated by variables at the absorption site, including possible drug degradation and significant inter- and intrapatient differences in the rate and extent of absorption.
The variability in systemic drug absorption can be minimized to some extent by proper biopharmaceutical design of the dosage form to provide predictable and reliable drug therapy. The major advantage of intravenous administration is that the rate and extent of systemic drug input is carefully controlled.
In pharmacokinetics, the overall rate of drug absorption may be described as either a first-order or zero-order input process. Most pharmacokinetic models assume first-order absorption unless an assumption of zero-order absorption improves the model significantly or has been verified experimentally.
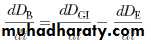
The rate of change in the amount of drug in the body, dD B/dt, is dependent on the relative rates of drug absorption and elimination. The net rate of drug accumulation in the body at any time is equal to the rate of drug absorption less the rate of drug elimination, regardless of whether absorption is zero-order or first-order.
Where D GI is amount of drug in the gastrointestinal tract and D E is amount of drug eliminated.
A plasma level–time curve showing drug absorption and elimination rate processes is given in:- During the absorption phase of a plasma level–time curve, the rate of drug absorption is greater than the rate of drug elimination. Note that during the absorption phase, elimination occurs whenever drug is present in the plasma, even though absorption predominates
At the peak drug concentration in the plasma the rate of drug absorption just equals the rate of drug elimination, and there is no net change in the amount of drug in the body.
Immediately after the time of peak drug absorption, some drug may still be at the absorption site (ie, in the GI tract or other site of administration). However, the rate of drug elimination at this time is faster than the rate of absorption, as represented by the postabsorption phase.
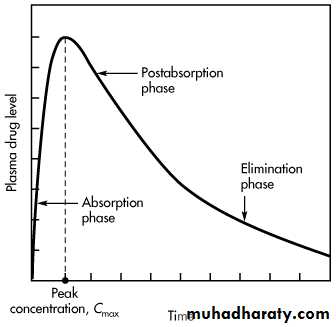
Plasma level–time curve for a drug given in a single oral dose
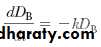
When the drug at the absorption site becomes depleted, the rate of drug absorption approaches zero, or dD GI/dt = 0. The plasma level–time curve (now the elimination phase) then represents only the elimination of drug from the body, usually a first-order process. Therefore, during the elimination phase the rate of change in the amount of drug in the body is described as a first-order process,
where k is the first-order elimination rate constant.
Zero-Order Absorption ModelZero-order drug absorption from the dosing site into the plasma usually occurs when either the drug is absorbed by a saturable process or a zero-order controlled-release delivery system is used. In this model, drug in the gastrointestinal tract, D GI, is absorbed systemically at a constant rate, k 0. Drug is simultaneously and immediately eliminated from the body by a first-order rate process defined by a first-order rate constant, k. This model is analogous to that of the administration of a drug by intravenous infusion

The rate of first-order elimination at any time is equal to D Bk. The rate of input is simply k 0. Therefore, the net change per unit time in the body can be expressed as:
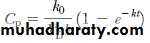
Integration of this equation with substitution of V DC p for D B produces
The rate of drug absorption is constant until the amount of drug in the gut, D GI, is depleted. The time for complete drug absorption to occur is equal to D GI/k 0. After this time, the drug is no longer available for absorption from the gut, and Equation no longer holds. The drug concentration in the plasma subsequently declines in accordance with a first-order elimination rate process.
First-Order Absorption Model
Although zero-order absorption can occur, absorption is usually assumed to be a first-order process. This model assumes a first-order input across the gut wall and first-order elimination from the body.
This model applies mostly to the oral absorption of drugs in solution or rapidly dissolving dosage (immediate release) forms such as tablets, capsules, and suppositories. In addition, drugs given by intramuscular or subcutaneous aqueous injections may also be described using a first-order process.

In the case of a drug given orally, the dosage form first disintegrates if it is given as a solid, then the drug dissolves into the fluids of the GI tract. Only drug in solution is absorbed into the body. The rate of disappearance of drug from the gastrointestinal tract is described by
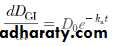
where k a is the first-order absorption rate constant from the GI tract, F is the fraction absorbed, and D GI is the amount of drug in solution in the GI tract at any time t. Integration of the differential equation gives
where D 0 is the dose of the drug.
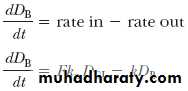
The rate of drug elimination is described by a first-order rate process for most drugs and is equal to –kD B. The rate of drug change in the body, dD B/dt, is therefore the rate of drug in, minus the rate of drug out:
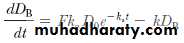
where F is the fraction of drug absorbed systemically. Since the drug in the gastrointestinal tract also follows a first-order decline (ie, the drug is absorbed across the gastrointestinal wall), the amount of drug in the gastrointestinal tract at any time t is equal to D 0e –k at .
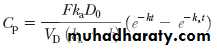
The value of F may vary from 1 for a fully absorbed drug to 0 for a drug that is completely unabsorbed. This equation can be integrated to give the general oral absorption equation for calculation of the drug concentration (C p) in the plasma at any time t, as shown below.
Equation shows that C max is directly proportional to the dose of drug given (D 0) and the fraction of drug absorbed (F).
A typical plot of the concentration of drug in the body after a single oral dose is presented in .
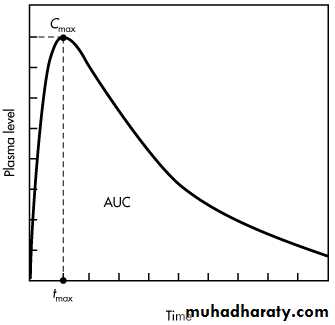

The maximum plasma concentration after oral dosing is C max, and the time needed to reach maximum concentration is t max. The t max is independent of dose and is dependent on the rate constants for absorption (k a) and elimination (k). At C max, sometimes called peak concentration, the rate of drug absorbed is equal to the rate of drug eliminated. Therefore, the net rate of concentration change is equal to zero. At C max, the rate of concentration change can be obtained by differentiating as follows
As shown in Equation, the time for maximum drug concentration, t max, is dependent only on the rate constants k a and k while C max is directly proportional to the dose of drug given (D 0) and the fraction of drug absorbed (F).
Calculation of t max and C max is usually necessary, since direct measurement of the maximum drug concentration may not be possible due to improper timing of the serum samples.
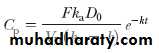
The first-order elimination rate constant may be determined from the elimination phase of the plasma level–time curve. At later time intervals, when drug absorption has been completed, ie, e –k at ≈ 0, Equation reduces to
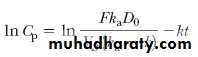
Taking the natural logarithm of this expression,
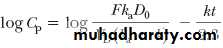
Substitution of common logarithms gives
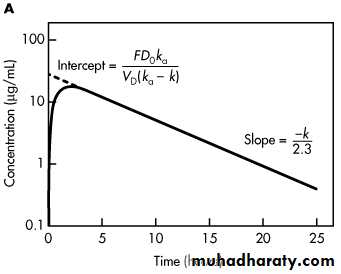
With this equation, a graph constructed by plotting log C p versus time will yield a straight line with a slope of –k/2.3
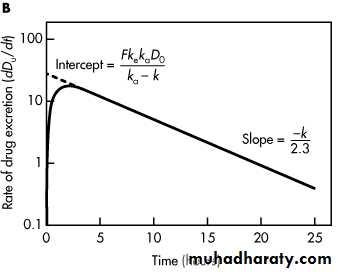
A. Plasma drug concentration versus time, single oral dose. B. Rate of urinary drug excretion versus time, single oral dose.
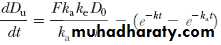
With a similar approach, urinary drug excretion data may also be used for calculation of the first-order elimination rate constant. The rate of drug excretion after a single oral dose of drug is given by
where dD u/dt = rate of urinary drug excretion, k e = first-order renal excretion constant, and F = fraction of dose absorbed.
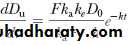
A graph constructed by plotting dD u/dt versus time will yield a curve identical in appearance to the plasma level–time curve for the drug. After drug absorption is virtually complete, –e–kat approaches zero, and Equation reduces to
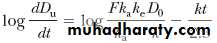
Taking the natural logarithm of both sides of this expression and substituting for common logarithms, Equation becomes
When log (dD u/dt) is plotted against time, a graph of a straight line is obtained with a slope of –k/2.3. Because the rate of urinary drug excretion, dD u/dt, cannot be determined directly for any given time point, an average rate of urinary drug excretion is obtained, and this value is plotted against the midpoint of the collection period for each urine sample.
Lag Time
In some individuals, absorption of drug after a single oral dose does not start immediately, due to such physiologic factors as stomach-emptying time and intestinal motility. The time delay prior to the commencement of first-order drug absorption is known as lag time.
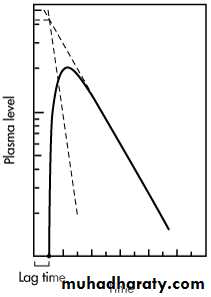
The lag time for a drug may be observed if the two residual lines obtained by feathering the oral absorption plasma level–time curve intersect at a point greater than t = 0 on the x axis. The time at the point of intersection on the x axis is the lag time.
The lag time can be determined graphically if the two residual lines obtained by feathering the plasma level–time curve intersect at a point where t > 0.
The lag time, t 0, represents the beginning of drug absorption and should not be confused with the pharmacologic term onset time, which represents latency, eg, the time required for the drug to reach minimum effective concentration.
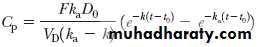
Two equations can adequately describe the curve in . In one, the lag time t 0 is subtracted from each time point, as shown in Equation.
where Fk aD 0/V D(k a–k) is the y value at the point of intersection of the residual lines in .
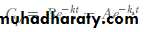
The second expression that describes the curve in omits the lag time, as follows:
where A and B represents the intercepts on the y axis after extrapolation of the residual lines for absorption and elimination, respectively.
Significance of Absorption Rate Constants
The overall rate of systemic drug absorption from an orally administered solid dosage form encompasses many individual rate processes, including dissolution of the drug, GI motility, blood flow, and transport of the drug across the capillary membranes and into the systemic circulation. The rate of drug absorption represents the net result of all these processes. The selection of a model with either first-order or zero-order absorption is generally empirical.
The actual drug absorption process may be zero-order, first-order, or a combination of rate processes that is not easily quantitated. For many immediate-release dosage forms, the absorption process is first-order due to the physical nature of drug diffusion. For certain controlled-release drug products, the rate of drug absorption may be more appropriately described by a zero-order rate constant.
The calculation of k a is useful in designing a multiple-dosage regimen. Knowledge of the k a and k allows for the prediction of peak and trough plasma drug concentrations following multiple dosing. In bioequivalence studies, drug products are given in chemically equivalent (ie, pharmaceutical equivalents) doses, and the respective rates of systemic absorption may not differ markedly. Therefore, for these studies, t max, or time of peak drug concentration, can be very useful in comparing the respective rates of absorption of a drug from chemically equivalent drug products.