

Haematology
Dr Khudhair Abass AliCollege of Medicine – Baghdad University
Objevtives
HaemostasisPlatelets
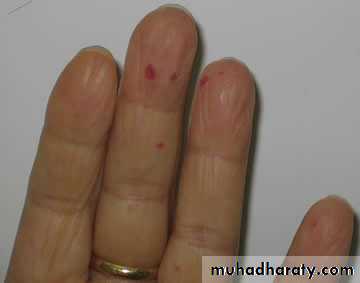
Idiopathic Thrombocytopenic Purpura (ITP)
Haemostasis
Mechanism : 1. Immediate response : Vasoconst. + platlets plug.2. Secondary : Fibrin deposition .
Stage 1 : Pre-injury :
. Vascular endoth. Produce substances prevent adhesion of plat. & WBC to vessel wall (NO2 , prostocyclin)Stage 2 : Early Haemostasis :
. Platlets Adherence & coagulation activation → Thrombin
Stage 3 : Fibrin clot formation .
Stage 4 : Limiting clot formation by natural anticoagulants (antithrombin , TFPI , protein C & S ).
Stage 5 : Fibrinolysis . Plasmin degrades fibrin → vessel recanalisation & tissue repair .
Lecture (1)
Clotting Factors : Half life
I. Fibrinogen 4-5 days
II. Thrombin 3 days
III. Tissue F. -------
IV. Ca++ -------
V. Labile F. 12-13 h
VI. No longer used -------
VII. Proconvertin 4-7 h
VIII. AHF 8-10 h
IX. Christmas F 1 day
X. Stuart power F 2 days
XI. Plasma Thromboplastin antecedent 2-3 days
XII. Hagman F 1 day
XIII. Fibrin stabilisig F. 8 days
All produce by the liver , FV also by plat. & endoth. Cells .
VIT. K dependent factors II VII IX X (inhibited by WARFARIN)
Blood Coagulation :
• 1. Extrinsic pathway : initiated by F VII interacting with Tissue Factor.• 2. Intrinsic pathway : initiated after exposure of subendoth. Collagen after vessel damage . ( XII XI IX VIII)
• Extrinsic Pathway Intrinsic Pathway
• TF XIa
• VIIa IXa
• VIIIa
• Xa
• Va
• Prothrombin Thrombin
• Fibrinogen Fibrin Monomer
• XIIIa
• Fibrin polymer clot
Platelets :
. Formed in Bone marrow from Megakaryocytes.
. Thrombopoiten (liver) stimulate formation & maturation of plat.
. Circulate in blood for 8-10 days before destroyed in RES.
. 30% pooled in spleen.
. Discoid 2-4 mm in diameter.
. Invaginated surface membrane to form Canaliculi.
Screening tests for bleeding disorders
1. Plat. Count (normal = 150-400 X 109 /L )
2. Bleeding time (normal < 8 min.), prolonged in thrombocytopenia, abnormal Plat.fun. , VonWolbrand disease, vascular & connective tissue diseases.
3. Prothrombin time (PT) : assess extrinsic pathway (normal 9-12 s). F II, V, VII, X
4. Partial thromboplastin time (PTT) : assess intrinsic pathway (normal 26-36 s). F II, V, VIII, IX, X, XI, XII, sever fibrinogen def. ,heparin, inhibitors.
5. Prolonged PT & PTT = common pathway (X, V, II & I) , or > one factor def.
6. Fibrinogen conc. (normal 1.5-4 g/L) , hypofibrinogemia e.g. liver dis. DIC.
7. Mixing tests with normal plasma differentiate between coag. Factor def. & presence of inhibitors (the prolonged time is not corrected).
8. plat. Functions tests.
Bleeding Disorders : suspect if
1. Multiple sites2. Spontaneous
3. Prolonged
1. Vascular purpura
a. Hereditary : Defective collagen .
b. Acquired : Immune, Inflammatory .
2. Platelets defects : Quantity, Quality
3. Coagulation defects :
Reduced synthesis of Fs
Abnormal Factors
Consumption: (DIC)
Inhibitors
4. Fibrinolytic defects :
e.g. Excessive fibrinolysis following therepeutic thrombolysis
Clinical assessment
1. Site of bleeding
Muscles, Joints, Retroperitoneal : indicate coagulation defect.
Purpura, Epistaxis, G.I.T., Menorrhagia: indicate low plat, plat. Funct.defect,
WWD.
Recurrent in single site – local abnormality
2. Duration
3. Precipitating Fs : Spontaneous or traumatic.
4. Surgery: Exo, Tonsilectomy, Circumcision.
Immediate : Primary Haemostasis
Delayed : Coagulation defect.
5. F.H. : Absence does not exclude hereditary dis.
6. Drugs .
Differentiation bet. Plat. dis & Coagul. dis
Plat dis. Coagul. dis.Sex F > M M > F
Age 20-40 y childhood
Onset Immediate Delayed
Type Mucocut. Deep Ts (M,J).
Local pr. Effective Not
Ex. ITP Haemophilia
Platelets Disorders
1. Thrombocytopenia < 150 X 109 / L
2. Thrombocytosis > 400 X 109 / L
3. Functional.
Thrmbocytopenia :
Spont. Bleeding – plat.count = < 20 X 109 / L< 10 X 109 /L – Risk of intracranial bleeding
Plat. Transfusion : indicated in
1. Bone marrow failure < 10 X 109 / L
2. Serious Haemorrhage.
Causes of thrombocytopenia :
1. Reduced production : Bone marrow dis.Aplastic Anaemia, Leukemia, Cytotoxics
B12 , Folic ac. def.
2. Increased Consumption :
ITP , DIC, TTP .
Non Thrombocytopenic purpura
1. Senile2. Factitious (self induced).
3. Henoch shoenlein purpura .
4. Vasculitis
5. Paraproteinaemia
6. Steroids .
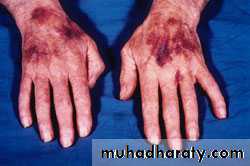
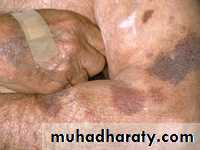
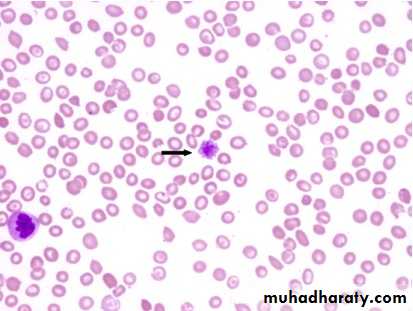
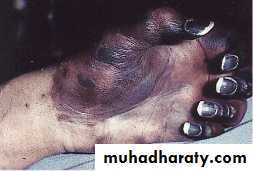
Idiopathic Thrombocytopenic Purpura (ITP)
Aetiology : Auto Abs against plat. memb. Glucoprotein Ilb / IIIa leads topremature removal by RES. May be assoc. with CTD, HIV, Pregnancy,
Drugs.
Clinical : Depends on degree of thrombocytopenia.
Spont. Bl. < 20 X 109 / L
< 10 X 109 / L – Risk of I.C. Bleeding
Higher count : Easy Bruising. Epistaxis, Menorrhagia.
> 50 X 109 / L – accidental diagnosis.
F > M
Preceding Viral inf. – unusual.
CTD at presentation or later
Splenomegally – Unusual.
Lab. : BI. Film – reduced plat. Count.
B. M. – Incr. or normal Megakaryocytes.
Plat. Abs - positive
D.D. : Acute leukemia, Aplastic An., DIC, SLE.
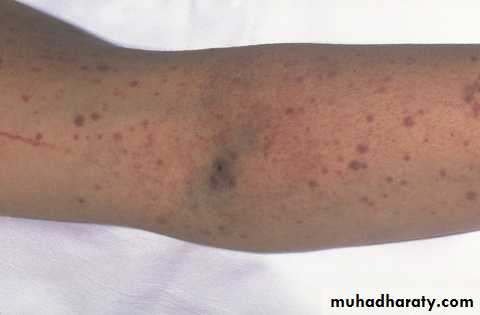
Management of ITP :
. Stable, plat > 30 X 109 / L – No treatment, except with Surgery or Biopsy. Spont. Bleeding – Prednisolone 1 mg / kg / day ( suppress Ab. Prod. and
inhibit phagocytosis) ,count will increases to normal or less in 2-4 weeks, then
reduce dose gradually.
. Relapse – Reintroduce Steroids
. Immunoglobulin i.v. (Block Ab receptor on RES) + steroids in severe
haemostatic Failure
. Anti-D i.v. (Bind RBC and saturate Ab. Receptors in Rh + ve (+ no splenectomy)
. Persistent or life threatening Bleeding needs plat. Transfusion
. 2 Relapses or Pri. Refractory to treatment needs splenectomy.
70% complete remission.
20-25% - improvement
5-10% require further treat. e.g. low dose steroids, Immunosup(Rituximab,
Ciclosporin)
summary
.All coagulation Fs produced by the liver.
Vit K dependent Fs : II VII IX X.
Plat count: 150 - 400x 109 /L
Bleeding Time (BT): < 8min
PT: 9-12sec (Asses Intrinsic Fs II ,V, VII, X)
PTT: 26-36sec ( Asses Extrinsic Fs II, V, VIII, XI, X, XI, XII)
Common pathway: Fs X, V, II, I (PT & PTT prolonged)
Suspect bleeding disorder : Multiple sites, Spontaneous, Prolonged)
ITP: Plat. < 10 X109 /L is symptomatic (risk of IC bleeding).
10-50 X 109 /L is symptomatic (spontaneous and easy bruising)
>50X109 /L : Accidental diagnosis
Splenomegally is unusual.
Objectives Lecture 2
1.Thrombotic thrombocytopenic purpura.2.Thrombocytosis.
3.Platelets function disorders.
4.Vessel wall abnormalities.
Hereditory haemorrhagic telengiectasia.
Henoch shonlein purpura.
Thrombotic Thrombocytopenic Purpura (TTP) (HUS)
Aetiology : Autoimmune disorder, mediated by Abs. against ADAMTS – 13 enzymes .
(This enz. normally clears VWF multimers to produce normal functional units and its deficiency results in large VWF multimers which cross – link platelets and leads to microvascular occlusion by plat. Thrombi e.g. brain, kidney).
Idiopathic, associated with drugs (Ticlopidine, Ciclosporine), HIV, Malignancy.
Clinically : Sporadic, Fluctuating, Familial: reported, F > M , young – adolescent
1. Fever
2. Anemia : Microangiopathic haemolytic (Fragmented RBCs) ,
↑ Reticulocyte count, Jaundice, ↑ LDH, -ve coomb’s test.
3. Thrombocytopenia – Purpura, count usually < 20 X 109 /L
4. Neurological: Convulsion, Coma, Paralysis ( Transient or prolonged) ,
Psychological .
5. Renal : intermittent proteinuria, Renal Failure (more in HUS)
Lecture (2)
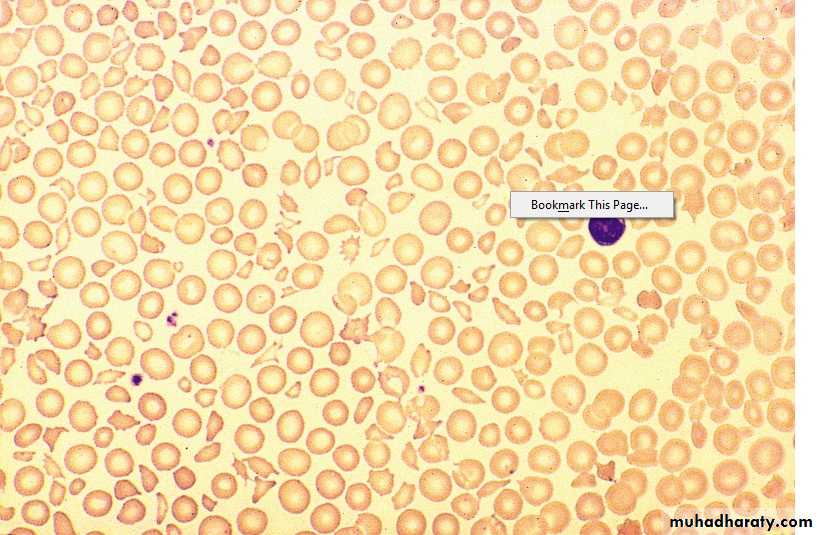
Lab. ↑ WBC, ↓ plat.
PT, PTT ------- NormalB.M. ------- Normal or increase cellularity
Gingival Biopsy – Helpful
DD : ITP
Prognosis : 10% Recurrent.
Untreated – 90% Mortality in 10 daysTreated – Mortality 20-30% at 6 months
Sudden death due to cardiac Micro thrombi
Treatment : Plasmapharesis with FFP 12 bags/day for weeks – Months
Corticosteroids, Rituximab have a role.Pregnant – May needs termination if no response
Plat. Function disorders
1. Acquireda. Iatrogenic
Aspirin – Cyclo – Oxygenase inhibitor
Clopidogrel – ADP receptor inhibitor
Abciximab – Gp 11 b / 111 a inhibitor
Tirofiban – Gp 11 b / 111 a inhibitor
Dipyridamol – phosphodi esterase inhibitor
b. Antibiotics: Penicillin, Cephalosporines
c. Heparin
d. CRF
e. Myelofibrosis
f. Paraproteinaemia
g. CTD
2. Congenital
a. Glanzmann’s thromboasthenia - Def. of GP 11b / 111a
AR, bleeding variable, often severe.
b. Bernard – Soulier dis. def. of GP 1b. AR
c. Defective plat. Granules – deficiency of dense granules
(b & c cause mild bleeding e.g. after trauma or surgery, rarely spont.)
Management :
Local mechanical pressure .Antifibrinolytic e.g. Tranexamic acid may be useful .
Plat. Transf. in severe bleeding.
Recombinant FVIIa in resistant bleeding in Glanzmann's.
VESSEL WALL ABNORMALITIES
1. Hereditary: HHT, EDD, Osteogenesis imperfecta2. Inflammatory: Bacterial, Viral, Vasculitis.
3. Abn. Collage: Senile, Scurvy, Steroids.
4. Metabolic : Liver disease, Renal, Amyloidosis.
5. Others: Factitious, Paraproteinaemia, Drugs.
Hereditary Haemorrhagic Telengiectesia (HHT)
(Osler – Rendu – Weber synd.)
Clinically := A.D., Telengiectasia and small aneurysms on Fingers, lips, Face,
Tongue, Nasal passages, Lungs, G.I.T. / Usually appear at adulthood.
Signif. No. have Pulmonary. A.V. malformation (PAVMS) → Arterial
Hypoxaemia due to Rt. To It. Shunt → paradoxical embolism →
stroke or cerebral abscess.
Recurrent Bleeding e.g. Epistaxis or Malaena → Fe def. An.
Treatment : Deficult because. Of multiple Bleeding sites.
Regular Fe therapy
Local Cautary or Laser, Oestrogens ?
Thrombocytosis
1. Reactive (most Common) 2. Clonala. Chr. Infl. Dis. E.g. ulc. Collitis a. Pri. Thrombocythaemia (ET)
b. CTD e.g. Rh. Arthritis. b. PRV
c. Malignancy e.g. Hodgkin’s dis. c. Myelofibrosis.
d. Acute Haemolysis d. CML.
e. Fe. Def. An. e. MDS.
f. GI bleeding
g. Post – splenectomy
Clinically :
Those of the underlying dis.
Haemostasis rarely affected
PRV, ET, Myelofibrosis, may present with thrombosis and rarely bleeding
Reactive : No splenomegally.
Ehlers – Donalos Dis :
AD. , Collagen defectFragile Bl. Vessel. And organ membrane. → Bleeding and organ rupture
Joint Hypermobility
Scurvy
. Vitamin C def. → affect normal synthesis of collagen. Perifollicular and patechial haemorrhage, sub- periosteal bleeding
. Diagnosis : Dietary History
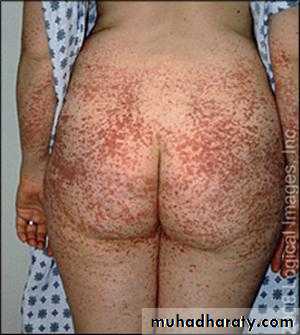
Henoch – Schonlein Purpura (HSP)
. Children and young adults .. Immune complex deposition in small vessel. With vasculities .
Often preceded by upper resp. inf.
Clinically :
1. Purpura: (raised) Buttocks and legs
2. Abdominal colicky pain and bleeding
3. Arthritis : Knees, Ankles .
4. Nephritis : 40% (May be later – 4 wks.)
Diagnosis : Clinical, Tissue biopsy ( IgA deposition in bl. Vessel.)
Prognosis : good, Adverse if HTN, RF, Proteinuria > 1.5 g / day
Treatment : Nonspecific
Steroids for joint or GI involvement
Nephritis – pulse iv steroids ± immunosuppressors.
Summary
TTPAutoimmune.
Clinically: 1.Fever
2.Microangiopathic haemolytic An.(Fragmented RBC)
3.Low plat.
4.Neurological.
5.Renal.
R: FFP + Plasmapharesis.
Thrombocytosis: Reactive or clonal.
Platelet function disorders: Acquired or congenital.
Vessel wall abnormalities : HHT, HSP
Objectives Lecture 3
Coagulation disorders;1.Congenital:
Haemophelia
Christmas disease
Vov-wilbrand disease
2.Acquired:
Disseminated intravascular coagulation
COAGULATION DISORDERS
Congental :
X-linked : Haemophelia. A and B
Autosomal : VWD, F II V VII X XI XII def.
Combined II VII IX X def.
Combined V and VIII
Hypofibrogenaemia
Dysfibrinogenaemia
Acquired
Under prod.: Liver failure.
Incr. Consumption: DIC
Immune – mediated: Acquired Haemophelia & VWD.
Others: Acquired F X def. (in amyloid)
Drug – induced
Inhibition Of function : Heparin
Inhibition Of synthesis : Warfarin
Lecture (3)
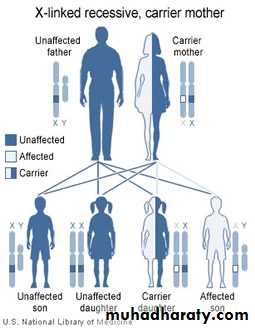
Haemophilia A :
. X-linked Rec.Haemophilic Male → 100% carrier daughters , boys all normal
Carrier Female → 50% carrier female, 50% Haemophilic male
. Antenetal diagnosis : chorionic villous sampling
. Similar severity in same family
. Female carrier : 50% have low F VIII , Mild bleeding ?
Clinically :
Severe Moderate Mild
F VIII < 1% 2-10% 10-15%
Spont. Bl. Mild Trauma Major Trauma
or surgery or surgery
. Recurrent bleeding into large joints (Knee, Elbow, Ankle, Hip)
Muscles (Calf, Psoas)
If no early treatment : Hot, swollen, very painful joint.
Rec. bleeding into joints → synov. Hypertrophy, cartilage destruction with OA .
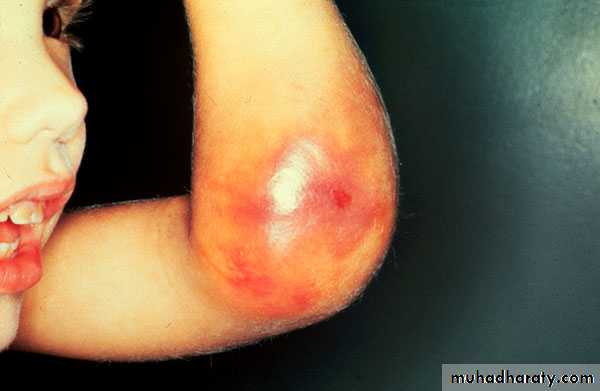
Muscles Hematoma : depend on site
Psoas muscle → compress fem. NCalf → compartment synd. → Ischemia , necrosis , fibrosis,
contracture and shortening of Achilles tendon.
Lab. ↑ PTT, Normal PT, plat. Count. & bleeding time , ↓ F VIII , -ve inhibitors.
Manag. : Avoid Trauma.
. Severe : 1- ↑ F VIII: (Donar Plasma or Recombinant) iv infusion F VIII concentrate.
2- Rest of bleeding site
3- If bleeding settled → Mobilization & physiotherapy
4- Hepatitis A,B immunization
5- Prophylactic: F VIII 2-3 X /wk – (at home) - expensive
. Mild : DDAVP (Desamino – des – aspartate – arginine vasopressin)
iv, SC , Nasal. It ↑ F VIII &VWF X 4, it covers minor surgery e.g. Exo
Complications :
1- HIV , HBV , HCV (< 1986)
2- Anti F VIII Abs. ( inhibitors ) 20%
3- OA & deformities .
Haemophilia B (Christmas dis)
↓F IX
X – linked
Clincally similar to A
Treatment : F IX concentrate
Inhibitors < 1%
Von Willebrand Dis. (VWD)
AD , M > F / most common bleeding disorder , usually mild.Function of VWF : 1- Carrier for F VIII (↓ VWF → ↓ F VIII).
2- Forms bridges bet. plat. & sub endoth. → plat. Function.
Clinically : different severity in same family , bleeding similar to plat. Dysf.
Bruising , Epistaxis , Menorrhagia, G.I. bleeding.
severe bleeding: ± after trauma or surgery.
Lab. : ↑ BT, ↑PTT, normal PT , ↓ VWF, secondary ↓ F VIII.
Manag. : Mild bleeding : local means or DDAVP , Tranexamic ac. may be useful
in mucosal bleeding.
Severe or persistent bl. : selected F VIII concentrate (contain F VIII +
VWF ).
Aquired Bleeding Disorders :
Disseminated Intravascular Coagulation (DIC)
Definition : Acq. Thrombohemorrhagic disorder, characterise by systemic activation of coagulation (Thrombosis) , with simultaneous coagulation Fs. (Notably F V & VIII) , plat. & fibrinogin consumption causing bleeding.
Lysis of fibrin →fibrin degredation products ( FDP ) including D – dimers.
Causes :
1- Infection ( sepsis ).2- Trauma.
3- Obstetric (Amniotic fluid embolism, placental abruption, pre-eclampsia).
4- Severe liver failure.
5- Malignancy.
6- Tissue destruction (Pancreatitis, burn).
7- Toxic / immulog. (ABO incompatab. , snake bite).
Clinically :
, 1- Signs of Thrombosis: Jaundice, blue toes, fingers gangrene, delirium,coma, respiratory failure, oliguria.
2- Signs of bleeding: S.C. bleeding, I.C. bleeding, Epistaxis, Gingival,
vaginal, renal .
Lab.
Anemia ( Microangiopathic Haemolytic ).
↑ PT & PTT
↓ Fibrinogen
↑ Bilirubin, ↓ plat. , FDP +ve
Manag.: Treat. Of underlying cause.
Deal with acidosis, dehydration, RF, Hypoxia.
FFP, cryoppt., plat. – if bleeding, or cover intervention (not roteinly)
Heparin – prophylactic doses (unless contraindicated) to treat
established thrombosis.
Liver diseases : ↓ synthesis of coagulation Fs.
↓ clearance of plasminogin activators.↓ plat. (Hypersplenism)
↓ vit K absorp. (cholestatic jaundice) → ↓ F II VII IX X
Lab. ↑ PT , ↑ PTT (in severe cases).
Treatment FFP , Plat. Vit. K
Renal Failure : Bleeding proportional to Urea concentration.
Similar to Plat. Dysf. Type (GIT is common).
Causes : An. , ↓ plat. , waste products.
Treatment Dialysis, plat. transf., Bl. , cryo. , DDAVP.
Vit K deficiency :
Sources : Green vegetables , intestinal flora.
Causes : ↓ intake, malabsorp., biliary obst. , antibiotics.
Clinical : plat. dysf. type of bleeding.
Lab : ↑ PT, ↑ PTT (in severe cases).
Treatment : Vit. K , im or iv for 3 days + treat. Of underlying cause.
Summary
Haemophelia:
X-linked
F VIII def.
Similar severity in the same family
Bleeding mainly into Joints and Muscles
Mild, Moderate ,Severe.
Complications: Inhibitors, Synovial hypertrophy, Cartilage destruction OA, Hepatitis, HIV.
Lab: ↑ PTT ,↓ F VIII
R: FVIII
VWD: AD
Different severity in the same family
Bleeding : platelets type
Lab: ↑ Bleeding time , ↑ PTT
R: Mild DDAVP
Severe: F VIII Concentrate
DIC: Common causes: septicemia, obstetrics, malignancy
Clinically: Bleeding and or thrombosis
Lab: ↑ PT & PTT, ↓ Fibrinogen, ↓platelets, ↑FDP
R: Underlying cause ± Heparin, FFP.
OBJECTIVES LECTURE 4
Deep vein thrombosis
Anticoagulation
Warfarin & Heparin
Antiphospholipid Syndrome
Thrombotic Disorders
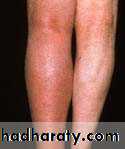
Deep Vein Thrombosis: (DVT)Mortality: 1-3%
Predisposing Factors:
1. Patients Factors: ↑Age, Obesity, Varicose veins, Previous DVT, F.H. Pregnancy, CCP, Puerperium.
2. Surgical: Major surgery, ( > 30 min ) , abdom., pelvic or lower limb ortho..
3. Medical: M.I., H.F., Inflm. bowel dis., Neph. Synd., Pneumonia, Stroke.
4. Hematology :PRV, ET, Deficiency of anticoagulants (antithrombin, prot. C,
prot. S), PNH, FV Leiden, Prothrombin gene G20210A .
5. Antiphospholipid synd.
Clinically :
Lower limb DVT : Typically Unilat. (may be bilat.).
Pain, swelling, warm, tender, dilated superficial Vs..
Examine thigh for DVT (proximal Involv.) → ↑ risk of PE.
Lecture (4)
Dif. Diagnosis :
1. Spontaneous or traumatic calf M. tear (sudden).2. Ruptured Baker cyst (Rh. Arthritis).
3. Infective cellulites (well demarcated swelling).
Investigations :
1. D-dimer.
2. compression ultrasound (doppler), sensitivity 99.5% in prox. (poplit. &
above), ↓ sensitivity & specificity for calf Vs.
3. Contrast venography – rarely used.
Management :
1. Elevation & Analgesia.
2. Thrombolysis for limb threatening DVT.
3. Main treat. : Anticoagulation : Heparin for at least 5 days, start warfarin
while on heparin which should be continued until INR in target range for
2 succsessive days (2-3, average 2.5).
If strong contraindication to anticoag. Or continue to have PE → IVC filter.
Warfarin – 6 wks to 6 Ms (or longer).
Recurrence : 2-3% /y in temporary risk, 10% in unprovoked DVT.
Post – thrombotic synd.: Damage to ven. valves by thrombus → persistent
leg swelling, heaviness & discoloration, sometimes ulceration.Patient PT
X international sensitivity indexـــــــــــــــــــــــــــــــــــ INR =
Normal control
Indications for Anticoagulation
Heparin Warfarin1. Prev. & treat. of VTE. 1. Prev. & treat. of VTE.
2. Precut. Coronary interv. 2. Art. Embolism.
3. Post thrombolysis MI . 3. AF with stroke risk.
4. Unstable Angina pectoris. 4. Mobile mural thrombus.
5. Non Q wave MI. 5. Extensive Ant. MI.
6. Acute periph. Art. Occlusion. 6. Dilated cardiomyopathy.
7. Cardio pul. By pass. 7. Cardioversion, Isch. Stroke . APS
8. MR & MS + AF, Rec. ven. Throm.
mechanical prosth. H. valves
Heparin : potentiate antithrombin anticoagulant. activity .
1.Unfractioned( UFH) .
2.Low molecular wt.(LMWH).
Advantage of LMWH :
1. 100% bioavailability =reliable dose dependent anticoagulation, no need for monitoring, except in very thin pt. & renal failure.
2. Once/day S C.
3. ↓ Osteoporosis & Thrombocytopenia.
Dose :
1.UFH :
Therapeutic : 5000 u iv followed by 20u/Kg/h by continuous iv inf.
Assess by PTT(Target 1.5-2.5 the control).
Prophylactic : 5000 u X 2 / S.C.
2. LMWH :
a. Enoxoparin :
Therapeutic : 150 u / Kg /S.C. daily.
Prophylactic : 2000 – 4000 u /S.C. daily.
b. Tinzaparin :
Therapeutic : 175 u / Kg/ S.C. daily.
Prophylactic 3500 - 4500 u/ S.C. daily.
Complications :
1. Bleeding : Treatment : Stop Heparin is usually enough (short half life)
If necessary protamine sulphate (Anti-dote).
2. Thrombocytopenia :
a. Early : First 24 hours (benign, due to plat. Clumping).b. Late : 5 – 14 days after treatment (anti-heparin/PF4 Abs. , bind to &
activate plat. & prothrombotic state causing thrombocytopenia.
Clinically : Asymtomatic – thrombosis in 50% , poor prognosis.
Manag. : Stop Heparin give lepirudin.
3. Osteoporosis (Heparin > 6 Ms ) .
Warfarin : inhibit vit K depend. Fs (II VII IX X).Take 3 days for anticoagulation.
Drug interaction
1. Potentiate : Sulphonamides, Cimetidine, Allopurinol, Metronidazole,
Thyroxin, Aspirin, Antibiotics.
2. Inhibition : Barbiturates, Rifampicin, CCP.
Dose : 10 mg on 1st & 2nd day , 5 mg 3rd day, & then according to INR.
Complications :
Bleeding : 1% fatal (intracranial)
Manag.: Look for the cause,e.g.drug interaction or↑dose. Stop warfarin temporarily, send for INR (may be enough) .
Minor : 1-2.5mg vit. K iv.
Major : 5-10 mg vit. K iv. slowly + FFP ( 15-30 ml / kg)
Prolonged INR with no bleeding : Stop or reduce the next Dose, small dose vit. K orally or iv. 1-2.5 mg. 8
Prophylaxis of ven. Thrombosis
Moderate risk High risk
1. Major surgery ( > 40 y + risk F) 1. Major abd. Or pelvic surgery for malig.
or with history of DVT or known
thrombophilia.
2. Major medical illness e.g. 2. Major Hip or Knee surgery.
HF, MI, Sepsis, Active malig. , 3. Neurosurgery
Neph. Synd. , Stroke + Paralysis
Methods
1. Mechanical : intermit. Pneumatic compr. , /feet pump, compression stoking. 2. Pharm. : LMWH, UFH, Warfarin, Aspirin.Contraindications
1. Recent surgery
2. Pre-Existing Haemorrhagic state ( liver dis. , Hemophilia )
3. Peptic ulcer.
4. Recent cerebral Hemorrhage.
5. Uncontrolled HTN
6. Frequent falls in old age.
Antiphospholipid Syndrome (AS)
Acquired prethrombotic tendency, in which arterial & or 0r venous thrombosis associated with finding in the plasma of lupus anticoagulant (LA) & or seropositivity for antiphosphoslipid Ab., most commonly Anticardiolipin Ab. (ACA)Clinically
1. Primary APS
2.Secondary APS is associated with : SLE, Rh.arthritis, Systemic sclerosis,
Behcets dis,Temporal arteritis.
Criteria A : Clinical:
1.Vacular thrombosis, one or more episode of arterial or venous or small
vessel thrombosis in any tissue/ organ.
2.Pregnancy morbidity: a.One or more unexplained death >10 wk gestation.
b.3 or more unexplained spont. Abortion <10 wk gestation.
c.One or more premature birth <34 wk gestation.
B :Lab:
1.Anticardiolipin Ab. On 2 or more occasions, 6 Wks apart.
2.Lupus anticoagulant Ab. On 2 or more occasions, 6Wks apart.
3.Prolonged PTT.
Diagnosis : At least ONE clinical & ONE lab. Criteria
Treatment:
Arterial thrombosis( stroke): Warfarin.
Recurrent VTE :Long term anticoagulation
Pregnancy :Heparin + Aspirin (Increase chance of successful pregnancy).
Summary
DVT :Aetiology: Surgery, Pregnancy, CCP (Hematological PRV,ET)
Hereditary, APS.
Clinically; unilateral, pain, swelling, tenderness
Lab: D-dimer, doppler, venography?
R: anticoagulation, How long?
APS: Thrombosis + LAB Criteria.
Objectives Lecture 5Blood transfusion: A Safe blood transfusion
BMT : types PRV ; A cause of polycythaemia.
Blood Products & transfusion
Donation :1.Whole Blood .
2. Apheresis e.g. platelets.
Blood components :
1.Red cell concentrate: (most of plasma removed ). ABO &Rh compatab. required.
Indication: Acute blood loss replac.& severe An.
2.Plat. Concentrate: ABO compatibility preferable.
Indication: Thrombocytopenia, Plat. Dysfunction.
3.FFP (150- 300 ml): ABO compat. recommended. Dose: 15 ml / Kg.
Indication.: Replacement. Of coagul. Fs def. , TTP .
4.Cryoprecipitate: (10-12ml pack): Contain fibrinogen(150-300mg) & coagulation
Fs. ( Each pack contain 80- 120u FVIII & VWF )
Indication: hypo- Fibrinogenemia , VWD ,Haemophelia.
Lecture (5)
Plasma Derivatives :
1.Coaguiation Fs : F VIII , F IX , recently: recombinant DNA technology.
2.Immunoglobulins : IV IgG (Ab.def., ITP, Gullian Bar. Synd.)
Prophylaxis of Varicella zooster.
Anti Rhesus D Ig
3.Human albumin :
5% :colloid fluid ( Crystalloid sol. Cheaper)
20% :Hypoprot. Odema (Nephrotic synd.), Ascitis in chr.
liver dis.
ABO Blood Groups :
Group Red cell A or B Ag Abs. in plasma % UKO None Anti A & B 46
A A Anti B 42
B B Anti A 9
AB A &B None 3
Donar Blood
Test for HIV ,HBV ,SYPHILIS ,ABO &RhRed cells FFP Pooled plat. --
Storage 40C - 30C 220 C
35 d 1 yr 5 d (agitated)
Adverse effects
Death: o.5/ 100 0001%: Symptoms e.g. fever , itch ,urticaria (repeated transfusions) .
Any symptoms or signs _take it seriously__WARNING !
1. ABO incompatibility :
Recep.IgM (Anti-A ,Anti-B or Anti AB + Transfused red cells
Activate complement system
intravascular haemolysis, hypotension, RF ,peribronchial edema & smooth muscle contraction2. Other immunological complication e.g. Transf. associated lung injury.
, 3. Infections: HBV, HCV, HIV, less than 1/1000 000,(bact. Contam. of plat.
Packs)
Conclusion: avoid unnecesary transfusion.
Signs & Symptoms of acute transfusion reaction :
(Fever, Chills, Tachycardia, Hyper or Hypotension, Collapse , Rigors, Flushing, Urticaria, bone, M., Chest & abd. Pain, SOB, Nausea , Resp.distress )Stop Transfusion
Temp, PR, BP, patient identity, Bl.pack, check compatibility form
1. Reaction only Fever or urticaria↓ ↓
FEVER URTICARIA
*Febrile Non Haemolytic Transf. Reaction * Mild Allergic Reaction
Treatment: Paracetamol Chlorpheneramine 10mg iv
Restart Transf. after 15-30min.at slower rate under observation if no more complications
Suspect ABO incompatibility
! Wrong Bl. Transfusion !YES NO
ABO incompat. Severe Allergic Reaction
Pack & giving set Blood Bank Bronch. Spasm,Angiodema,Abd.pain
i.v.Saline, Urine Output(catheter) Hypoten.
U.O.<100ml/h: give Frusemide ↓
Treat DIC NO YES
Bact. Contam. Stop Transf.
Pack & Giving Set back Chlorphen. 10 mg i.v.
to Bl.Bank, Bl.Culture, O2,Salbutamol Neb.
Repeat Bl.group & comp. Adrenaline 0.5 mg i.m. in
FBC, Coagul.screen ,Bioch. severe Hypot.or Bronchosp.
GUE. Send pack &giving set to Bl.
Broad spectrum antibiotics. Bank.
O2, Fluid.
.
No Bact. Contam.
If acute Dyspnoea. Hypot. Bl. Gases, CXR, CVP, Pul.cap. Pr.
↓ ↓
High CVP Normal CVP
Fluid Overload Transf. related Acute Lung Injury
O2, Frusemide 40-80 mg (Typically within 6h )
SOB, cough, CXR: bilat. nodular infilt.
Stop Transf. 100% O2 ,
Treat as RDS, Ventilation if necessary.
Bone Marrow & peripheral stem cell Transplant (BMT)
Allogenic :Stem cell from Donar, given i.v. to patient( previously conditionedby chemo ± radioth. → engraft into pt.BM (function 3-4 wks).
Sources :
1. Related (HLA identical siblings ).
2. Unrelated ( closely HLA matched Volunteer ) .
Indications :
1.Neoplastic disorders (Leukemia, AML, ALL, CML, MDS, Myelofibrosis )
2. Failure of haemostasis e.g.( Aplastic an.)
3.Inherited Defects e.g.( Thalassemia ) .
Complications : Mucositis, infection, bleeding, pneumonitis, infertility, Chr.GVHD.
secondary malignant dis.
20 yr. +HLA identical subling.> Best results : pt. with minimal residual dis. .Age
25 % die from procedure ,GVHD,
Long survival :50% for leukemia.
GVHD :
Aetiology: cytotoxic activity of donor T lymphocytes ,which become
sensitized to their new host,regarding it as foreign. This may cause
acute or chr. GVHD .
Acute: Ist 100 d, one third of pt. Mild – Lethal, → Skin rash, jaundice,
diarrhea.
Prevention: HLA matching + Immunosupp.drugs
Chronic :May follow acute or independent, later than acute, resemble CTD.
Reduced intensity BMT :↓ mortality
↓ Intensive conditioning
↓ Toxic , old pt.
↑ relapse ?
Autologous :
Pt. own stem cell, from Bl. Or BM ,harvested & frozen → conditioning→reinfuse .
Indications : Dis. do not primarily involving hematological Tissues, or
Pt. who had achieved good Remission.
e.g. AML CR2, Myeloma, poor –risk Hodgkin, High- grade Non-Hodgkin.
Marrow recovery : 2-3 wks .
No risk of GVHD.
↓ mortality .
↑ Recurrence .
Stem cells treated to remove any residual leukemic cells is still
investigated.
Polycythaemia
0.52 %< 180 g / L (18 g /dl) in M PCV < Hb
% 0.48 < 165 g/ L (16.5 g /dl) in F PCV
Classification : RBC Mass Plasma Vol
True Increased Normal
Relative Normal Decreased
Relative : e.g. Dehydration, Diuretics, Alcohol.
True : Primary Secondary Myeloprplif. Disorders ↑EPO (Hypoxia) Inappropriate ↑
( PRV) ↑ Altitude Renal dis.
Lung dis. CHD, Hepatoma
↑ Affinity Hb Bronchogenic Ca.
Uterine fibroid.
Phaeochromocytoma.
Cerebellar
haemangioblastoma
Polycythemia Rubra Vera
Age: more than 40y.Clinically :
1. Incidental.
2. Symptomatic :Hyperviscocity : Lassitude, loss of conc., headache, dizziness,
blackouts, pruritis (worse after hot bath) , epistaxis, peripheral arterial dis.
e.g. CVA (thrombosis) , venous thromboembolism,.
Peptic ulcer is common (sometimes bleeding ) .
Plethoric face , majority has palpable spleen .
Diagnosis: ↑ RC mass , ↑PCV , ↑ Plat.& Neutrophil (often) , JAK -2 Mut.(97%)
Hypercellular B.M. + Absence of secondary causes .
Treatment : ↓ risk of thrombosis : Venesection 400-500 ml (elderly less) every
5-7 wks → PCV 45%- & MAINTAIN AT THIS LEVEL .
Hydroxycarbamide ( Hydroxyurea), or ᾱ interferon → suppress marrow prolif.
& ↓ transform. Into myelofibrosis , control spleen size .
RA P 32 In old pt.
Prognosis : Survival 10- 20 y with Treatment. CVA & IHD 60%
Conversion to other myeloprolif. Dis. 15% , Acute leukemia ( RA P)
Summary
Blood Transfusion: Life saving proceedure,not always safe. has to be done properly, from right donor to the right recipient under medical supervision. .Most common side effects are febrile & urticarial reactions, which should be managed with suspension of transfusion for 15-30 minute, treat symptomaticaly & observation for progression of signs & symptoms. Restart if no progression.
BMT: Allogenic, Autologous.
PRV : Clonal disease , could be asymptomatic, or hyperviscocity
Symptoms.
Spleen palpable 50%
Lab: ↑ Hb, PCV., Red cell mass, WBC. Platelets
R: venesection
Hydroxyurea
Objectives:
WBC differential, functions,↑↓Lymphadenopathy.
Splenomegally
WBC DISORDERS
⁄ L * Normal Count : 4 –10 X 10 9 * Lecoerythroblastic picture : presence of immature WBC & ERYTHROBLASTin peripheral Bl.
* Peripheral bl. Contain Neutrophils (N) ,Lymphocytes (L),Eosinophils (E),
Basophils (B) ,Monocytes(M) .
* Myelocytes & Metamyelocytes may appear in peripheral Bl. in infection.
* Leucocytosis : ↑ Total WBC or only one type of cell (differential) .
* Leucopenia : ↓ Total WBC or only one type of cell . NEUTROPHILS : 2---7 X 10 9 ∕ L
Recognize ,ingest & destroy foreign particles, spend 6-10 h in circul.→ removed by spleen .
* Neutropenia: < 1.5 x 10 9 ∕ L ( Asymptomatic –Sepsis )
If < o.5 x 10 9 / L – Critical , Fever sore throat ,perianal pain, skin inflam. → SHOCK if no antibiotics .
Lecture (6)
Neutrophelia Neutropenia,
Infection: Bacterial ,Fungal Viral ,Bacterial( TF) , Protozoal( malaria)Trauma: Surgery ,Burn . CTD, Alcohol.
Infarction: MI, PE ,Sickle C.Crises. BM infilt.: Leukemia, MDS,
Inflam.: Gout, R.arth.,U.C. Drugs
Malignant: Solid Tumors, Hodgkin's.
Myeloprolif.: PRV,CML.
Physiological: Exercise, Pregnancy.
Drugs causing Neutropenia
Analgesia,Anti-infl.: Gold,Penicillamine. Anti-Thyroid : Carbimazole.
AntiArrhythmics: Quinidine . AntiHypert.: Captopril, Nifedipine.
AntiDepres.: Amitryptylline. Anti-Malarial: Chloroquine.
AntiConvulsants: Phenytoin, Carbim.
Antibiotics: Sulphonamide,Penicillin, Chloramphenicol.
Miscellaneous: Cimetidine,Ranitidine,Chlorpropamide,Zidovudine
Lymphocytes : 1.5- 4 x 10 9 / L
T Lymphocytes : Mediate cell immunity.
B Lymphocytes : Mediate humeral immunity.
Lymphocytosis Lymphopenia
Infection :Viral, Bacterial (Pertussis) Inflam.: CTD.Lymphoprplif.: CLL, Lymphoma. Lymphoma , Post- splenectomy R.F., Steroids, Severe comb.im.def. Cytotoxics.
Eosinophils : 0.04 -0.4 X10 9 / L
1. Intracellular killing of Protozoa &Helminthes ., 2.Allergic reaction.Eosinophilia :
1. Allergy : Hay fever, asthma, eczema.
2.Infection ;Parasitic.
3.Drug hypersensitivity: gold,sulphonamide.
4:Skin dis.
5:CTD. Polyarteritis nodosa,Hypersensitivity.
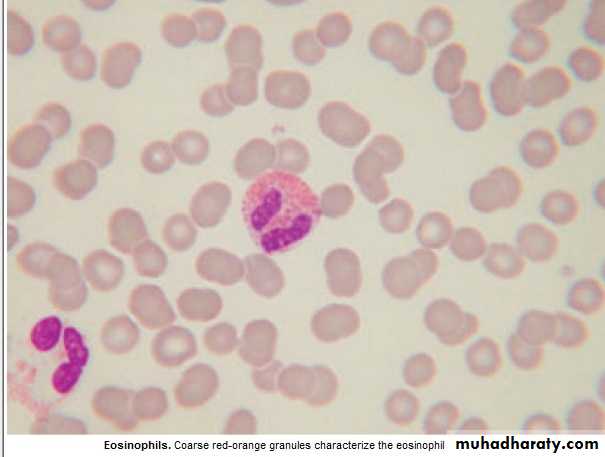
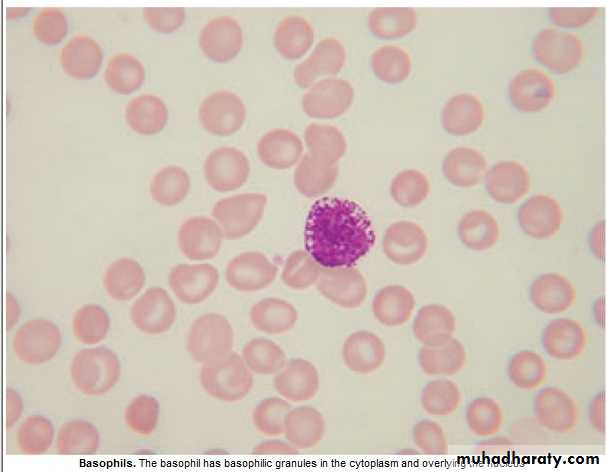
Basophils : 0.01 -0.1 x 10 9/ L
Involved in hypersensitivity reaction.Basophilia : 1. Myeloprolif.dis.: PRV, CML.
2. Inflam.: Acute hypersensitivity ,Ulcer. Colitis(UC).
3.Fe def. An.
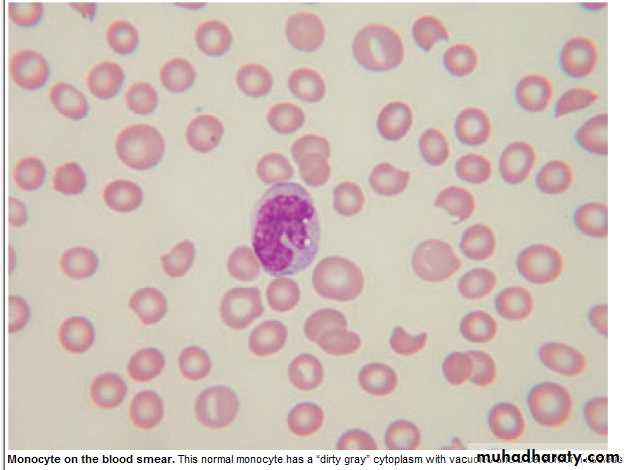
Monocytes : 0.2 – 0.8 x 10 9 / L
Migrate into tissue → Macrophage .
Monocytosis : 1. Infection (TB).
2.Inflam. : CTD, UC.
3.Malig.: Solid tumors'.
Lymphadenopathy
Causes : Infection : 1.Bacterial : strep. ,TB, brucellosis. Typhoid fever.2.Viral ;EBV, HIV.
3.Protoz. :Toxoplasmosis.
4. :Fungal :Histoplasmosis.
Neoplastic : 1. Primary : Lymphoma, Leukemia
2.Secondary : lung, breast, thyroid, stomach. CTD :Rh. Arthritis,SLE.
Sarcoidosis,Amyloidosis.
Drugs :Phenytoin.
History :Speed of onset,Pain,Associated symptoms,Wt. loss,Night sweat., Itching.
Exam.: Site,Localised or Generalized.
Consistency: Hard,Soft,Rubbery.
Area of drainage, + General exam.
Investigations : CBP ( ↑ Neutrophils = Inflam. ,Haematological dis.), ESR, CXR, Biopsy.
Splenomegally
Causes : 1.Congestive: 4.Haematological:
a.Intrahep. Portal. HT. a. Red cell disorders
Cirrhosis,Hepatic V. Throm. Megalob. An.,Hemoglobinopat.
b.Extrahep.Portal HT. b.Autoimmune haemolytic An.
c.Cardiac:CHF, Const.peric. c.Myeloprolif.disorders :CML,PRV,
2.Infective: Myelofibrosis,ET.
a.Bact.:Endocarditis,Septicemia,TF, d.Neoplastic:Leukemia, Lymphom.
b.Viral :Hepatitis,EBV,Cytomegal. 5.Other Malignancies:Metast.(rare)
c.Protoz.:Malaria,Kala-azar. 6.Gauchers dis.
d.Fungal:Histoplasmosis. 7.Miscellan.:Cysts,Amyl.Thyrotoxico.
3.Inflammatory/Granulomatous:
Feltys syndrome,SLE,Sarcoidosis.
Clinically: + Liver → Lympho. Or Myeloprolif.dis.,Liver dis.,Amyloid.
+ LN → LymphoProlif.
Abdom. discomfort+ backache, splen. Infarc.→ severe abd. Pain., rupture ?
Investing.: FBC (Pancytopenia =Hypersplenism ?) ,Abd. US & CT ,CXR (Mediast. LN )
BM ,Splenectomy if all negative.
Summary
Lymphadenopathy:Most common cause is infection: painful ,tender,examine
drainage area.
Neoplastic cause is usually painless, examine other areas,
spleen and liver.
Send for CBP.
Splenomegally:
Most common cause is infection.
Check for lymphadenopathy and hepatomegally.
Send for CBP.
Objectives L7
Hematological malignancies: Acute Leukemias:
Lymphoblastic(ALL),Myeloblastic(AML)
Chronic Leukemias:
Myeloblastic(CML),Lymphoblastic(CLL)Haematological Malignancies
Corruption of the processes controlling proliferation & apoptosis of bl. Cells.If mature cells involved → Chronic Leukemia or Low grade Lymphoma
Primitive cells involved → Acute Leukemia or High grade Lymphoma
Leukemias : 100/ 100 000 /y .Acute Leukemia ˂ 50 % M ˃ F ,
Acute: All ages , ALL ↑ in children, AML ↑ in Old.
Risk Factors : 1. Ionizing radiation
2. Cytotoxic drugs
3.Retroviruses.
4.Genetics
5.Immunological.
Lecture (7)
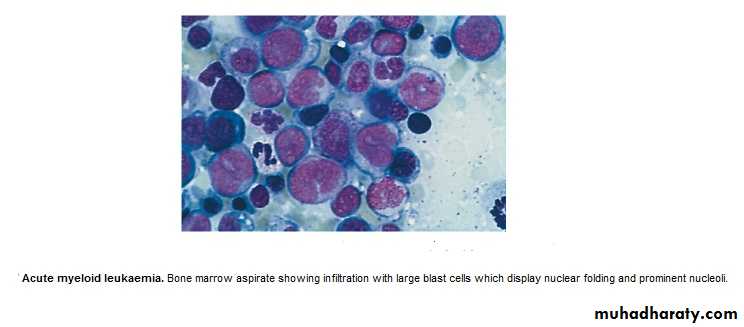
Acute Leukemia : ALL & AML
Failure of cell maturation, accumulation Of useless immature cells in B.M. on the
expense of normal haemopoeitic elements, with spilling of immature cells into Bl.
Clinical Features are: marrow failure ( Anemia, Bleeding, or Infection)
Investigations :
CBP : Anemia ,WBC : ↓ 1 X 10 9/ L --- ↑ 500 X 10 9/ L (Majority ˂ 100 X 10 9/L. Platelets : usually very low.
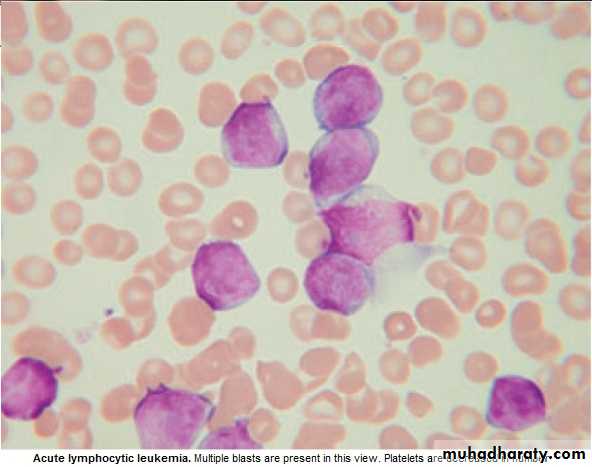
BLAST in Peripheral Blood .
BM : Confirm the diagnosis, replacement of normal elements by leukemic
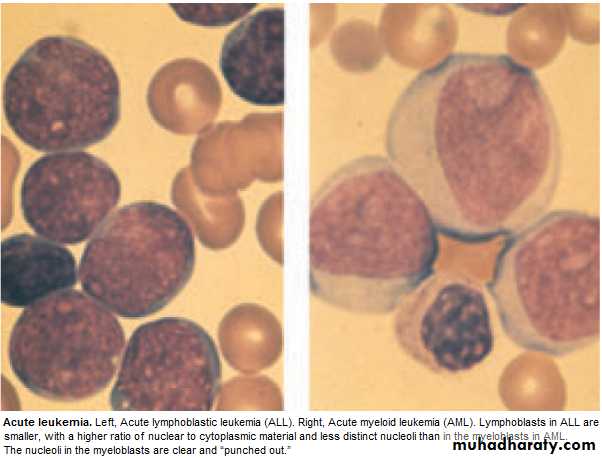
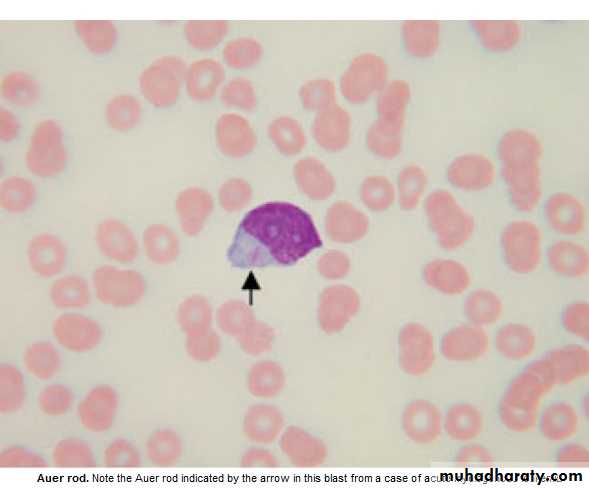
blast cells ( ˃ 20 % of the cells ),presence of Auer rods in the cytoplasm
of the blast cells = AML.
Management :
1. Decide whether to give specific treatment or not,(It is aggressive with ↑S.E.)
which may not be appropriate for elderly or patients with other serious dis.→ supportive treatment.
Specific Treatment :
Preparation for specific treat. :1.Identify & treat infection
2.Correct Anemia :Red cell Concentrate.
3.Thrombocytopenic bleeding ← Platelets'.
4.Hicman line (if possible)
5.Explain Therapeutic regimen to pt. & obtain consent.
-Aim of Treat. = destroy leukemic cells without destroying the normal stem cells
→Repopulation of haemopoeitic Ts.
Remission induction :Bulk of tumor is destroyed by combination chemotherapy
→Severe bone marrow hypoplasia (require intensive support.)
Remission consolidation :If Rem. Achieved → Residual dis. attacked by No. of courses of chemo.→ marrow hypoplasia (BMT may be included at this stage)
Rrmission maintenance :If still in Rem.(ALL) → Repeated cycles of Chemo. up to 3yr.
+ CNS prophylaxis = Intathecal (IT) Methotrexate + cranial radiation.
Failure of Rem. in induction. →Alternative combination of drugs.(poor prognosis)
Relapse during treat. Or after → poor prognosis.
Drugs commonly used in treat. Of Acute Leukaemia
phase ALL AML
Induction Vivcristine (iv) Daunorubicine(iv)
Prednisolone(po) Cytarabine(iv)
L-asparaginase(im) Etoposide(iv&po)
Daunorubicine(iv)
Methotrexate(IT)
Consolidation Daunorubicine(iv) Cytarabine(iv)
Cytarabine(iv) Amsacrine(iv)
Etoposide(iv) Mitoxantrone(iv)
Methotrexate(iv)
Maintenance Prednisolone(po)
Vincristine(iv)
Mercaotopurine(po)
Methotrexate(po)
Supportive treatment:
Anemia : Red Cell Concentrate Transfusion.Bleeding :Thrombocytopenic bleeding require platelets.Prphylactic plat.transf.to
maintain plat.˃ 10 X 10 9/L. Treat any Coagulation abnormalities.
Infection : Fever ˃ 38oC , ˃ 1h in Neutropenic pt. indicate possible septicemia. Start
Broad spectrum antibiotic(Gentamycin + piperacillin± Vancomycin),to continue for at least 3ds after the fever resolved.
Flucanazole for oral & pharyngeal moniliasis.
Amphotericin iv for systemic fungal inf.(candida or aspergillosis) for 3wks.
Herpes simplex and zoster : antiviral(aciclovir).
Isolation is debatable (psychological).
Metabolic problems :Monitor fluid, hepatic, renal function,anorexia & difficult drinking
→ iv fluid & electrolytes. Cellular breakdown during induction(tumor lyses'
syndrome) → ↑K, ↑Uric acid, ↑phosphate,& ↑Ca. → RF ?
Allopurinol + iv Hydration .
Psychological : Keep pt. informed ,answer question, allay fear as possible.
Prognosis : Median survival without treat.is 5wks….Few Ms with supportive treatment.
80% ˂ 60y age achieve remission with specific treat.
Survival depends on risk factors (chromosomal analysis)
5y survival for AML 21%-76%, ALL 20-37% .
Chronic Myeloid Leukemia ( CML )
* Myeloproliferative stem cell disorder resulting in prolif. Of all hematopoietic
Lineage but predominantly in the granulocytes series. Chiefly 30-80y, 20% of all
leukemia. * 95% have Philadelphia (ph ch) = shortened ch. 22 resulting from translocation
of material with ch 9.
* BCR(breakpoint cluster region on chr.22 + Fragment from chr. 9 carries ABL oncogen → BCR ABL Gene codes for protein with Tyrosine Kinas activity→ play a role in the dis. As an oncogen.
CML has 3 phases:
1.Chronic phase:. The dis. is responsive to treat., lasting 3-5y. With the introduction of imatinib → ˃ 5Y.
2.Accelerated phase :(not always seen), dis. control more difficult.
3.Blast crises:dis transforms into acute leukemia (AML%70 ,ALL30%),Refractory to
treat., It is the major cause of death. Prior to imatinib therapy, 10% CML→ AML/y, now only 0.4-2.5% after up to 5y treatment with imatinib.
ph. Ch. –ve CML: 0ld pt., ↑Male, ↓Plat. ↑ Monocyte,respond poorly to treat.
Median survival 1y.
Clinically: 25% - asymptomatic at diagnosis.
Tiredness 37% Anorexia 12%SOB 21% Abdom. Fullness 10%
Abdom. Pain 21% Bruising 7 %
Lethargy 13% Vague ill health 7%
*Splenomegally 90% (10% massive) ± friction rub(splenic infarction)
*Hepatomegally 50%
*LN Unusual.
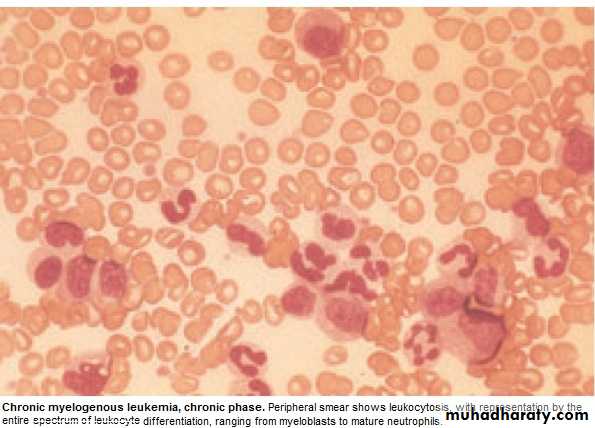
Investigations:1.FBC: An.(normochromic normocytic).WBC 10-600X10 9/L(full range
of granulocytes precursors from myeloblast to mature neutrophils,
myeloblast ˂ 10%, ↑Eosinoph .&Basophils.
Platelets: ↑ in ⅓ (up to 2000x109/L . Nucleated RBC are common.
Accelerated phase: ↑ % of more primitive cells.
Blast transformation: Dramatic ↑ Blast in circul., ↓plat.
Basophils ↑ with dis. Progress.
2. Bone marrow: to confirm diagnosis &phase of the dis.(morphology & chromosome analysis .
3. Bl. LDH & Uric acid ↑ ( ↑ cell breakdown)
Management:
Chronic phase:1.Imatinib inhibit BCR ABL Tyrosine Kinase(TK) activity & reduce
. the uncontrolled prolif.of WBC.. It is Ist line therapy in chronic phase
complete cytogenic response (disappearance of ph chr.) in 76% at 18 Ms of therapy.
Failure of response or progress on imatinib → 2nd generation TK Inh. e.g.
Dasatinib, or Nilotinib : 2. Allogenic BMT
3. Hyroxycarbamide (Hydroxyurea):still used for initial control of the dis.or
palliative treat.(no effect on ph chr. & on onset of blast transformation).
4. Interferon-ἀ : was Ist line of treat.± Ara-C: control CML in 70% of cases.
Accelerated phase: Pt. presented with this phase ,imatinib is indicated if not already
received. Hydroxyurea is also effective. Low dose cytarabine can be trie Blast Transform.: ALL response better than AML.( + supportive treat.)
Pts. Progressing to advanced phase on imatinib may respond to 2nd genera. TK inh. or BMT.
Chronic Lymphocytic Leukemia (CLL)
* Most common leukemia(30%).M:F is 2:1, median age at presentation 65-70y.* B lymphocytes fail to transform & produce Abs →Increasing mass of immuno-
incompetent cells → ↓immune function & normal BM haematopoeisis.
Clinically:
*insidious onset. 70% incidental diagnosis (routine FBC).
*Anemia, Infection, Painless Lymphadenopathy ± splenomegally & systemic symptoms e.g.Night sweat ,or wt. loss.
Investigations:
1.FBC: Mature lymphocytes ˃ 5x 10 9/L. characteristic morphology & markers, CD19 & CD23.
2.↑ Reticulocyte count & +ve coombs test.= haemolytic An.(may occur)
3.S. Igs: to assess the degree of immunosuppression(common & progressive).
4.B.M. exam. Is not essential for diagnosis, but helpful for prognosis(diffuse involve.= poor prognosis),& to monitor response to therapy.
Main prognostic factor is stage of the dis.,CD38 ,Mutation of IgVH Genes suggest poor prognosis.
Staging of CLL:
Stage A: 60%,No anemia, Normal Plat. Count, ˂ 3 areas of LN enlargement.
Stage B: 30%,No anemia, Normal Plat., 3 or more areas of LN enlarg.
Stage C : 10%,Anaemia & or ↓ Plat., regardless of the No. of areas of LN enlarg .
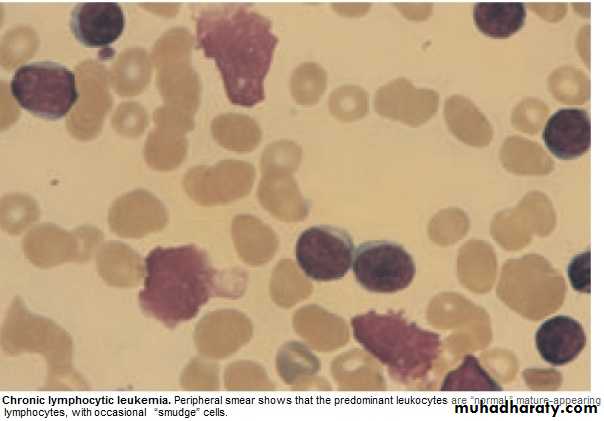
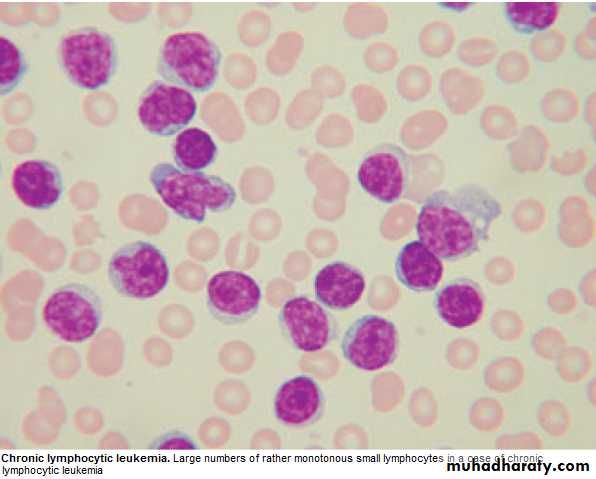
Management:
* No specific treat. For most stage A, unless progression, life expectancy is normal inmost patients.
*Treatment: Indications: 1. BM failure. 2. Massive or progressive LN PATHY or Splenomegally.
3. Systemic symptoms e.g. wt. loss or night sweat.
4. Rapidly ↑ lymphocyte count.
5. Autoimmune haemolytic An.,or ↓ Plat.
Stage B & C :Chlorambucil
Recently Fludarabine + Cyclophosphamide → ↑ rem.rates & dis. Free survival ( ↑ risk of infection )
B.M. Failure or autoimmune cytopenias → corticosteroid.
Supportive care: Symptomatic, Anemia or thrombocytopenia → Transfusion.
Treat. Of infect., Ig for hypogammaglobulinaemia.
Radiotherapy: LN causing discomfort or obstruction.& for symptomatic splenomegaly Splenectomy : may be , to improve low Bl. Count. due to autoimmune destruction or due to hypersplenism & can relieve massive splenomegally.
* Prognosis: Overall survival is 6y., stageC 2-3y, ,50% die from infection.
Rarely → Aggressive high grade Lymphoma(Richters syndrome).
Prolymphocytic Leukemia
* Variant of CLL, mainly in Male ˃ 60y. 25% T cell variety* Massive splenomegaly, + Little Lymphadenopathy.
* WBC often ˃ 400 x 10 9/ L (Characteristic cell is large lymphocyte with prominent nucleolus.
* Treat. : Is generally unsuccessful & prognosis very poor.
Leukapharesis, Splenectomy, & chemo. May be tried.
Hairy cell Leukemia :
* Rare,chronic lymphoproliferative B cell disorder, M 6:F 1 , median age 50y.* General ill health, & recurrent infection.
* Splenomegaly 90%, LN unusual.
* Lab.: Severe Neutropenia, Monocytosis,& characteristic HAIRY CELL in Bl. & B.M..
→ CD25 & CD103.
* Treat.: Cladribine & Deoxycoformycin.
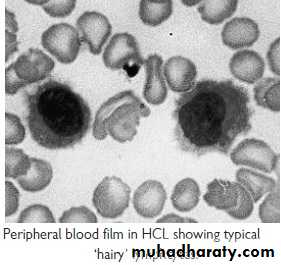
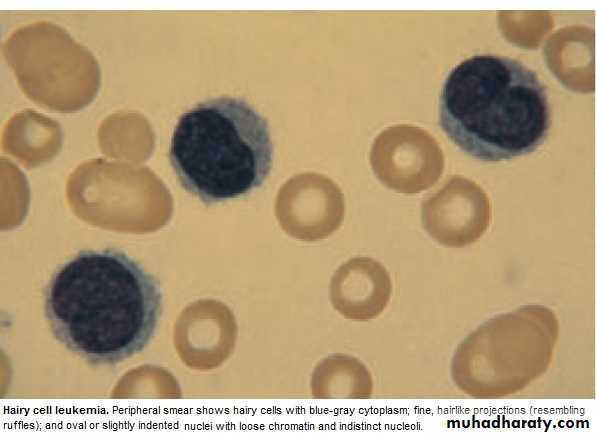
Summary
Acute leukemias: ALL & AMLFailure of maturation.
Lab: > 20% Blast.in BM.
R: Supportive or aggressive
CML:↑profil. Of all haemopo.cells.
90% +ve ph chr.
3 phases: Acute, Accelerated, Blast.
R:Imatinib
CLL: B Lymphocytes fail to transform & produce Ab.
3 stages.
R:Fludarabine
Objectives
LymphomasHodgkin
Non- Hodgkin
Paraproteinaemias
Lymphomas
* Neoplasm arise from lymphoid tissues. majority are of B cell origin. diagnosed from pathological changes on biopsy as Hodgkin or Non-Hodgkin lymphoma.
* Non-Hodgkin lymphomas are classified as:
1. High - grade: rapidly dividing, present for Wks.before diagnosis, life threatening.
2. Low - grade: slowly dividing, present for Mns.before diagnosis, indolent.
Hodgkin lymphoma (HL)
* Histological hallmark is Reed –Sternberg cells.
* 4/ 100 000/y, slight M. excess, median age 31y
Aetiology unknown, ↑in educated background & small families, X3 ↑ likely with
PMH of inf. Mononucleosis ,(no association with EBV).
* Pathological classification:
1.Nodular lymphocyte predominant,(5%),slow growing, localized, rarely fatal.
2.Classical HL:
a. Nodular sclerosing (70%), ↑in young F.
b.Mixed Cellularity (20%), ↑elderly.
• c. lymphocyte-rich: (5%) ↑M.
d.Lymphocyte –depleted : (rare)
Lecture (8)
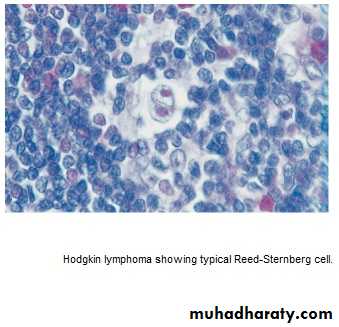
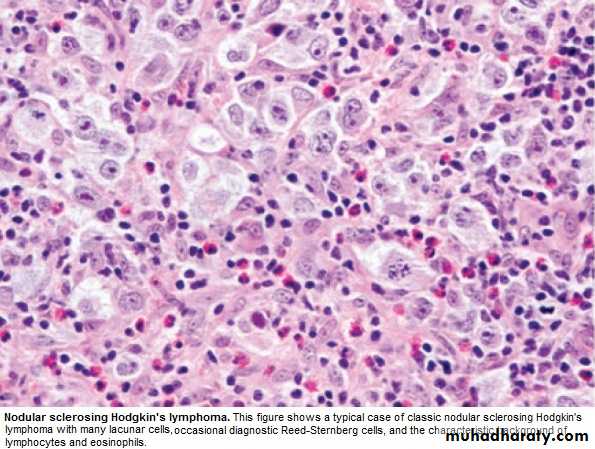
Clinically :
* PAINLESS,RUBBERY LN-pathy, ↑ neck,& supraclavicular. Young with Nod. Sclerosis may have large mediastinal masses (asymptomatic or dry cough & SOB).Isolated subdiaphragmatic nodes ˂ 10% at diagnosis.
* Hepatosplenomegaly may be present but not always indicate dis. In them.
* Extra nodal spread to bone brain or skin --- rare.
Clinical Stages: (Ann Arbor)
1.Stage I : Single LN region(I)or extra lymphatic site(IE).
2.Stage II: 2 or more LN regions(II),or an extralymph.site & LN regions on the same side of (above or below) the diaphragm(IIE).
3. Stage III: LN regions on both sides of the diaph.with(IIIE) or without (III) localized
extra lymph. Involv. Or involv. Of spleen(IIIs) or both (IIISE).
4. Stage IV: Diffuse involv. Of one or more extralymph. Tissue e.g. liver or BM.
Note: 1. Each stage is subclassified into :
A. No systemic symptoms.
B. Systemic symptoms ( Weight loss, drenching sweat)
2. Lymphatic structure = LN, Spleen, Thymus, Waldeyers ring, Appendix &
peyers patches.
Investigations:
* FBC: may be Normal. Anemia or Lymphopenia :poor prognostic F.. ± ↑ E , N & ESR.* Renal f. tests: (to insure normal function before treat.).
* Liver f. tests: Abnormal may reflect hepatic infilt., LN in porta hapatis → obs.j.
* LDH: ↑ = Poor prognostic F.
* CXR: Medast mass ?
* CT: chest, abd., pelvis → staging. Bulky dis.= ˃ 1o cm single node mass(poor prog.F) * LN biopsy:surgical or percut. Needle biopsy.
Management:
* Historically: stage IA & IIA : Local radiotherapy(Radio.T)
*.Recently: Early-stage dis.has better outcome if chemotherapy(Chemo.T) included.
Majority are now treated with Chemo,T.+ Radio.T.
ABVD regimen (doxorubicin,vinblastine,bleomycin & dacarbazine).
Early –stage: 4 courses ABVD,followed by radio.T. to involved LN.
Response by CT & positron emission tomography(PET).
S.E.of ABVD: doxorubicin → cardiac,bleomycin → pulmonary.
↓ MDS /AML & infertility.
Advanced stage: Chemo.T. alone. 6-8 cycles ABVD., Therapy resistant → BMT.
Prognosis:
Early- stage: 90% CR, majority cured.Advanced –stage: 45-70% cured.
Relapse within a year → good salvage rate with autologous BMT.
Relapse after 1y → Chemo.T
Non- Hodgkin lymphoma ( NHL )
* Monoclonal prolif. Of lymphoid cells of B cell (70%) or T cell (30%) origion.* 12 new cases / 100 000/y. slight ↑ M ,median age 65-70y.
Aetiology:
* Late manifest. Of HIV.
* EBV,HTLV.(Certain type of lymphoma).
* H pylori –Gastric lymphoma.
*Chromosomal lesion: t(14:18) –Follicular lymphoma.
*Immunosuppressed pts. after organ transplant, congen. immunodef states.
Clinically: The most important factor is grade:
* High grade: High proliferation rate, rapidly producing symptoms, fatal if untreated, potentially curable.
* Low grade: Low prolif. rate, may be asymptomatic for many Ms before presentation.
indolent course, not curable.
Other forms include : mantle cell L. &MALT lymphoma (less common).
Clinically:
* Often widely spread at presentation, including extra nodal sites.* LN-pathy + systemic upset: Weight loss, sweats, fever & itching. ± H.splenomegally.
* Extra nodal involves BM, gut, thyroid, lung, skin, testis, brain & bone.
BM involv. is more common in low grade (50-60%) than high grade (10%).
Compression syndrome: gut obst., Ascitis, SVC obst. & SC compression.
* Staging is same as HL, but NHL is more likely to be stage III & IV at presentation.
Investigations: as in HL + the followings:
1.Routine BM aspirate & trephine biopsy.
2.Immunophenotyping of surface antigens to distinguish T & B cell tumors.
3. Igs: some lymphomas are associated with IgG or IgM paraproteins( serve as
markers for treatment response.
4. Uric acid ↑ with treat..
5. HIV.
Management:
Low grade lymphoma:
* Asymptomatic: may not require treat.
Indications: marked systemic symptoms, LN causing discomfort or disfigurement.
* Options:
1. Radiotherapy: localized stage I (rare)
2. Chemotherapy: Chlorambucil (oral), well tolerated but not curative.
More intensive iv chemo. In young → better quality of life
but no survival benefit,
3.Monoclonal Ab. Therapy: target surface Ag.on tumor cells.→ cell apoptosis.
Rituximab(R)(anti-CD20) → durable response in 60%,when given alone.
Ist line therapy = R-CVP ( Cyclophosphamide, Vincristine, Prednisolone)
4.Relapse: ↑ dose Chemo. + BMT (Study is going on).
High grade lymphoma: needs treat. At initial presentation.
1. Chemotherapy: > 90%: CHOP(Cyclophosphamide, Doxorubicin,Vincristine,Predn)
2. Radiotherapy: Few stage I, residual bulky dis. After chemo, compression(e.g. SC.)
3.Monoclonal Ab. Therapy: R-CHOP → ↑ complete response rate & survival.
4.Autologous BMT: relapsed chemosensitive pt.
Prognosis:
Low grade: Indolent, remitting & relapsing,median survival- 10y.High grade: 80% respond initially to treat. But only 35% have dis.-free survival at 5y.
Paraproteinaemia:
Gammopathy: Overproduction of 1 or more Ig.( detected by electrophoresis).Polyclonal: Causes: Acute or chr. Inflamation e.g.Infections, Sarcoidosis, Autoimmune disorder, Malignancy.
Monoclonal:(=M Protein) occurs with N or ↓ other I gs.
Myeloma, Lymphoma, Amyloidosis, CTD e.g. Rh. arthritis ,HIV, Solid
tumors or no underlying dis. gammapathy of uncertain origion (MGUS):
Common condition with increasing age.
Paraprotein is present in Bl. but with no other features of MM, WM,Lymphoma,
or related dis.
Clinical: Usually asymptomatic, normal FBC & Biochemistry. Ig : mild ↑with
no immune paresis. BM → Plasma cells < 10% of nucleated cells.
Prognosis: After 20y → 25% MM or related disorder.
Annual monitoring.
Waldenstrom macroglobulinaemia
Low- grade lymphoplasmacytoid lymphoma associated with IgM paraprotein,causing clinical features of hyperviscocity syndrome. Rare, ↑ elderly, ↑ M.
Clinically: Nose bleed, bruising,confusion & visual disturbance.
May present with An.,systemic symptoms, splenomegaly,or LN-pathy.
Lab.: ↑ IgM + ↑ plasma viscocity.
BM: Infiltration with lymphoid cells & prominent mast cells.
Manag.:
Severe hyperviscocity & An. → plasmapheresis.
Chlorambucil: effective but slow.
Fludarabine: more effective.
Rituximab: also effective
Summary
Hodgkin Lymphoma: Histological ly: Reed –sternberg cells.High & Low grades.
Unknown etiology.
Painless LN ± Hepatosplenomegally.
R : ABVD.
Prognosis: Majority cured.
Non-Hodgkin Lymphoma:
High & Low grades.
High grade is potentially curable.
Often wide spread at presentation + systemic symptoms.
R : Low grade: R-CVP.
High grade:R-CHOP.
Objectives:
Multiple Myeloma.Myeloproliferative diseases:
Myelofibrosis.
Essential Thrombocythemia.
PRV\
Infectious mononucleosis.
Multiple Myeloma (MM) Lecture 9
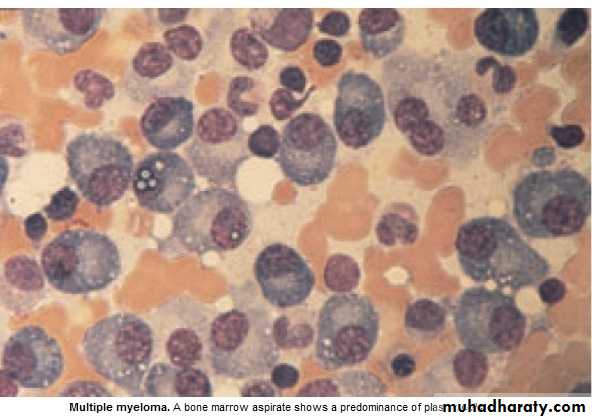
* Malignant prolif. Of plasma cells.
Normally: Plasma cell produce polyclonal Ig = variety of heavy chains are produced,
& each may be of kappa or lambda light chain type.
Myeloma: Plasma cells produce Ig of a single heavy & light chain (monoclonal prot-
ein = paraprotein), in some cases only light chain is produced →appear
in urine (Bence Jones proteinuria).
Classification:
Type of paraprotein %
IgG 55
IgA 21
Light chain only 22
Others(D,E, non-secretory) 2
*Majority of malignant plasma cells are present in BM ,small No. in circulation.
*Malignant plasma cells produce cytokines → stimulate osteoclast →bone reabso-
rption. → lytic lesions → pain, fracture, & ↑ Ca++.
*BM involv. → An. or pancytopenia.
Clinically:
4/100 000/y, M:F 2:I,Median age 60-70y.PATHOLOGY EFFECTS SYMPTOMS
Marrow involv. Bone erosion by osteoclast Pain
path. Fracture Severe local pain
↑Ca++ Lethargy,thirst.
BM failure → An. Tiredness
↑ paraprot.& L chain Renal damage Renal failure
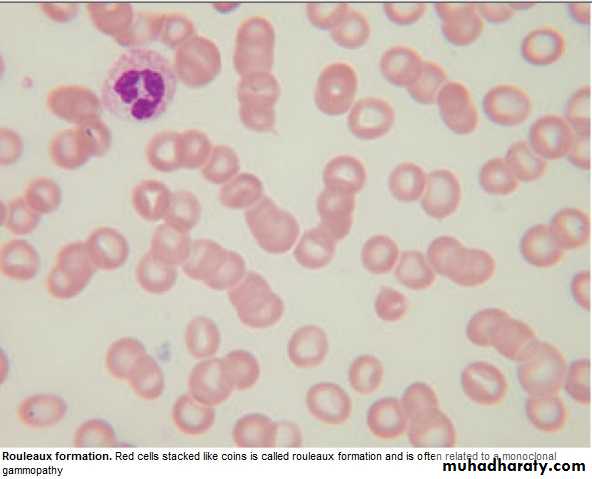
↑ viscocity Blurred vission, headach
Amyloidosis Nephrotic syndrome
↓ Normal plasma cells ↓ immunity ↑ infection(UTI,Resp.)
Diagnosis: Requires 2 of the followings:
1. BM malignant plasma cells >20% (BM aspiration)
2. S & or urinary paraprotein.(Bl & urine protein electrophoresis)
3. Skeletal lytic lesions.(X-rays/skeletal survey)
other investigations:
1. FBC: (Degree of BM failure),↑ ESR (not specific), urea & electrolytes, creatinine, uric ac., S.Ca++ & albumin.
coagulaton screen?, B2 microglobulin, MRI( SC compr.).
2.Bl. & urine immunoelectrophoresis(type of paraprotein).
3.Quantification of paraprotein,& other Igs( ↓ = immune paresis).
Management:
Asymptomatic: treat. may not be required.Immediate support:
1. High fluid intake to treat renal impairment & hyper Ca++.
2. Analgesia for bone pain.
3. Biphosphanates for hyperCa++(it also reduce bone pain & risk of fracture, may cause apoptosis of malignant plasma cells)
4.Allopurinol:prevent urate nephropathy.
5. Plasmapheresis for hyperviscocity.
Chemotherapy:
1. Old pts.: Ist line chemotherapy: Thalidomide + Melphalan + Prednisolone →
median survival 51 Ms. 2. Young Pts.: Ist line chemo. until max. response(plateau phase),then autologous
BMT (prolong survival but no cure).
3. Relapse: Bortezomib.
Radiotherapy:
For localized pain not responding to analgesia, pathological fracture & for emergency treat. Of SC compr. Complicating extradural plasmacytoma.
Prognosis: Poor prognostic features: ↑ B2-microglobulin, ↓ albumin, ↓Hb or ↑Ca++ at presentation. 5% survive 5y.
Myeloproliferative disorders:
Group of diseases characterized by clonal prolif. Of marrow erythroid precursors(PRV),
Megakaryocytes( Essential thrombocythaemia & Myelofibrosis) or myeloid cell CML), some having overlapping features,& often progression from one to another e.g. PRV to myelofibrosis.
MYELOFIBROSIS:
BM is initially hypercellular, with excess abnormal megakaryocytes which release
growth factors e.g. platelet-derived growth factor, to the marrow microenvironment
→ reactive prolif. of fibroblasts →marrow fibrosis.
Clinically: Age ↑ ˃ 50y, Lassitude, wt. loss, night sweat. SPLEEN MASSIVELY ENLARGED (extramedullary haematopoiesis).
Lab.: 1.FBC: Leukoerythroblastic An. ↑ reticulocyte count, tear drop RBC. WBC: ↓ - ↑ .
Platelets: ↑ , N, ↓, Giant form may be seen. 2. ↑uric ac., folate def. is common.
3. BM aspirate: difficult, Biopsy: Excess megakaryocytes,↑ reticulin & fibrous T. replac.
4. JAK -2 mutation 50%.
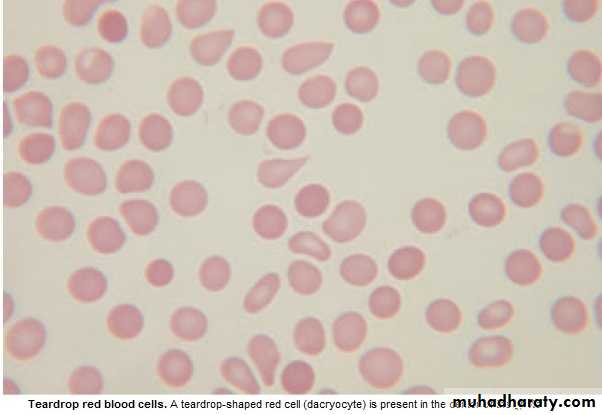
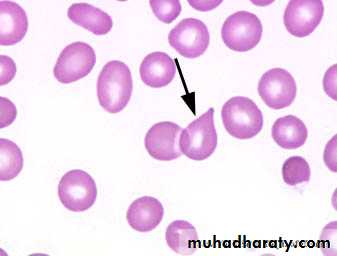
Management & prognosis:
Median survival: 4y.( range 1-20y).Treat.: Control symptoms:
RBC transf. for An.
Folic ac. To prevent def.
Hydroxycarbamide to control spleen size, WBC count, systemic symptoms.
Splenectomy: grossly enlarged, or pancytopenia(hypersplenism)
BMT: may be considered for young pt.
Essential thrombocythaemia;
Malignant prolif. Of megakaryocytes result in raised level of circulating platelets,that are often dysfunctional.
Reactive causes of ↑plat. must be excluded.
Clinically: Median age: 60y, may be asymptomatic with ↑ plat. Count.
Vascular occlusion or Bleeding.
Small % → Acute leukemia or Myelofibrosis.
Lab.: ↑ plat.,JAK-2mutation 50%
Treat.:
* ↓ risk pt.: ( age < 40y, plat.c. < 1000 x 10 9/L & no bleeding or thrombosis) →
may not require treat.
* Plat, c. > 1000 x 10 9/L, with symptoms, or with risk factors for thrombosis(DM,
HTN),→ Treat. to control plat.c../ e.g. Hydroxycarbamide or Anagrelide(inh. Of
megakaryocytes maturation). Radioactive phosphorus( P32) for old age.
* Aspirin for all pts. to reduce risk of thrombosis.
Infectious mononucleosis (IM)
*Acute viral illness, most often caused by EBV(CMV, HIV-1 & Toxoplasmosis may
cause similar clinical syndrome).acquired from asymptomatic excreters via saliva
by droplet infection or kissing.
Clinically: I.P.:Prolonged, undetermined.
Fever, Headache,& malaise,followed by severe pharyngitis (± tonsillar exudate, Ant. & Post. cervical LN-pathy), palatal petechiae, periorbital oedema,splenomegaly, inguinal & axillary LN. & macular, petechial,or erythema multiforme rashes may
occure. Fever resolved in 2 wks & fatigue in another 2 wks.
Death is rare : resp.obst., splenic rupture, thrombocytopenia, or encephalitis.
Investigations: 1. FBC: Atyical Lymphocytes in peripheral Bl.
2. Paul-Bunnel or Monospot test: detect heterophil Ab.present during acute illness
& convalescence (to be repeated if initially –ve).
3.Specific EBV serology (immunofluorescence) can be used to confirm the diagnosis
Complications:
Common Uncommon RareSevere pharyngeal Odema Neurological Ruptured spleen
Antibiotic-induced rash Cranial nerve palsies Resp. obstruction
(80-90% with Ampicillin) Polyneuritis Agranulocytosis
Prolonged post-viral fatigue Transverse myelitis Lymphoprplif. Dis.
Hepatitis(80%) Meningoencephalitis
Jaundice( <10%) Hematological Hemolytic An. Thrombocytopenia Renal Abnormal GUE Interstitial nephritis Cardiac
Myocarditis, abnormal ECG
Pericarditis
Management:
1.Symptomatic:2.If throat culture revealed B-haemolytic streptococci → penicillin.(avoid
ampicillin
& amoxicillin → macular rash .
3.Severe pharyng. Odema → prednisolone 30 mg 5d.
4.Antiviral: not effective.
5.Avoid contact sports (splenic rupture) until splenomegay has completely
resolved .
Summary
MM:IgG is the most common type.
Diagnosis 1. > 20% plasma cells in BM.
2.Serum or Urine paraprotein.
3.Skeletal lytic lesions.
R: Thalidomide + Melphalan+ Prednisolone
Myelofibrosis:
Massive splenomegally.
R: Symptomatic ± Hydroxycarbamide.
Splenectomy, BMT.
ET: ↑plat. clinically: vascular occlusion or bleeding.
R: Aspirin + Hydroxycarbamide when indicated.
Infectious mononucleosis: EBV, Atypical L, Paul-Bunnel,symptom.R.