Haematology
Dr Khudhair Abass AliCollege of Medicine – Baghdad University
Chronic Myeloid Leukemia ( CML )
* Myeloproliferative stem cell disorder resulting in prolif. Of all hematopoieticLineage but predominantly in the granulocytes series. Chiefly 30-80y, 20% of all
leukemia. * 95% have Philadelphia (ph ch) = shortened ch. 22 resulting from translocation
of material with ch 9.
* BCR(breakpoint cluster region on chr.22 + Fragment from chr. 9 carries ABL oncogen → BCR ABL Gene codes for protein with Tyrosine Kinas activity→ play a role in the dis. As an oncogen.
CML has 3 phases:
1.Chronic phase:. The dis. is responsive to treat., lasting 3-5y. With the introduction of imatinib → ˃ 5Y.
2.Accelerated phase :(not always seen), dis. control more difficult.
3.Blast crises:dis transforms into acute leukemia (AML%70 ,ALL30%),Refractory to
treat., It is the major cause of death. Prior to imatinib therapy, 10% CML→ AML/y, now only 0.4-2.5% after up to 5y treatment with imatinib.
ph. Ch. –ve CML: 0ld pt., ↑Male, ↓Plat. ↑ Monocyte,respond poorly to treat.
Median survival 1y.
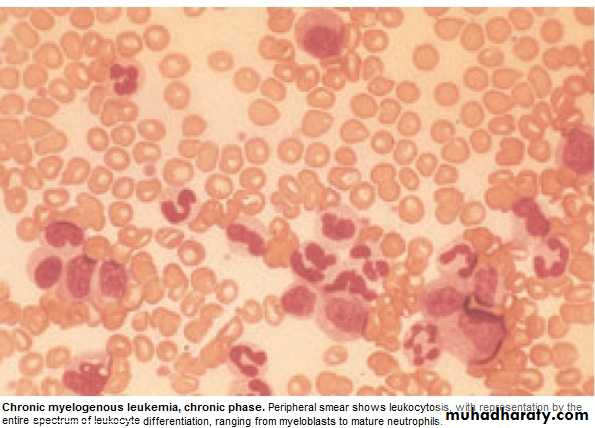
Clinically: 25% - asymptomatic at diagnosis.
Tiredness 37% Anorexia 12%SOB 21% Abdom. Fullness 10%
Abdom. Pain 21% Bruising 7 %
Lethargy 13% Vague ill health 7%
*Splenomegally 90% (10% massive) ± friction rub(splenic infarction)
*Hepatomegally 50%
*LN Unusual.
Investigations:1.FBC: An.(normochromic normocytic).WBC 10-600X10 9/L(full range
of granulocytes precursors from myeloblast to mature neutrophils,
myeloblast ˂ 10%, ↑Eosinoph .&Basophils.
Platelets: ↑ in ⅓ (up to 2000x109/L . Nucleated RBC are common.
Accelerated phase: ↑ % of more primitive cells.
Blast transformation: Dramatic ↑ Blast in circul., ↓plat.
Basophils ↑ with dis. Progress.
2. Bone marrow: to confirm diagnosis &phase of the dis.(morphology & chromosome analysis .
3. Bl. LDH & Uric acid ↑ ( ↑ cell breakdown)
Management:
Chronic phase:1.Imatinib inhibit BCR ABL Tyrosine Kinase(TK) activity & reduce
. the uncontrolled prolif.of WBC.. It is Ist line therapy in chronic phase
complete cytogenic response (disappearance of ph chr.) in 76% at 18 Ms of therapy.
Failure of response or progress on imatinib → 2nd generation TK Inh. e.g.
Dasatinib, or Nilotinib : 2. Allogenic BMT
3. Hyroxycarbamide (Hydroxyurea):still used for initial control of the dis.or
palliative treat.(no effect on ph chr. & on onset of blast transformation).
4. Interferon-ἀ : was Ist line of treat.± Ara-C: control CML in 70% of cases.
Accelerated phase: Pt. presented with this phase ,imatinib is indicated if not already
received. Hydroxyurea is also effective. Low dose cytarabine can be trie Blast Transform.: ALL response better than AML.( + supportive treat.)
Pts. Progressing to advanced phase on imatinib may respond to 2nd genera. TK inh. or BMT.
Chronic Lymphocytic Leukemia (CLL)
* Most common leukemia(30%).M:F is 2:1, median age at presentation 65-70y.* B lymphocytes fail to transform & produce Abs →Increasing mass of immuno-
incompetent cells → ↓immune function & normal BM haematopoeisis.
Clinically:
*insidious onset. 70% incidental diagnosis (routine FBC).
*Anemia, Infection, Painless Lymphadenopathy ± splenomegally & systemic symptoms e.g.Night sweat ,or wt. loss.
Investigations:
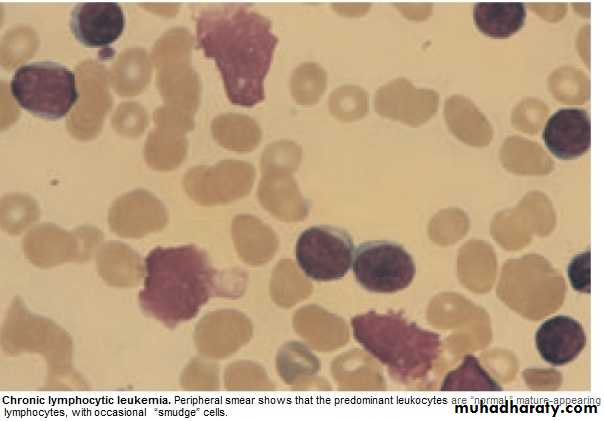
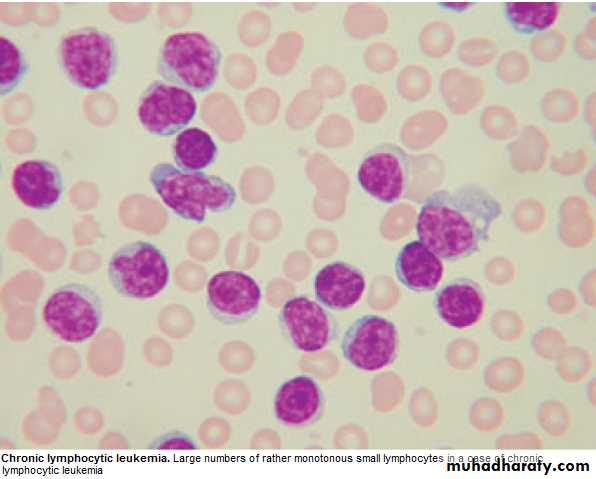
1.FBC: Mature lymphocytes ˃ 5x 10 9/L. characteristic morphology & markers, CD19 & CD23.
2.↑ Reticulocyte count & +ve coombs test.= haemolytic An.(may occur)
3.S. Igs: to assess the degree of immunosuppression(common & progressive).
4.B.M. exam. Is not essential for diagnosis, but helpful for prognosis(diffuse involve.= poor prognosis),& to monitor response to therapy.
Main prognostic factor is stage of the dis.,CD38 ,Mutation of IgVH Genes suggest poor prognosis.
Staging of CLL:
Stage A: 60%,No anemia, Normal Plat. Count, ˂ 3 areas of LN enlargement.
Stage B: 30%,No anemia, Normal Plat., 3 or more areas of LN enlarg.
Stage C : 10%,Anaemia & or ↓ Plat., regardless of the No. of areas of LN enlarg .
Management:
* No specific treat. For most stage A, unless progression, life expectancy is normal inmost patients.
*Treatment: Indications: 1. BM failure. 2. Massive or progressive LN PATHY or Splenomegally.
3. Systemic symptoms e.g. wt. loss or night sweat.
4. Rapidly ↑ lymphocyte count.
5. Autoimmune haemolytic An.,or ↓ Plat.
Stage B & C :Chlorambucil
Recently Fludarabine + Cyclophosphamide → ↑ rem.rates & dis. Free survival ( ↑ risk of infection )
B.M. Failure or autoimmune cytopenias → corticosteroid.
Supportive care: Symptomatic, Anemia or thrombocytopenia → Transfusion.
Treat. Of infect., Ig for hypogammaglobulinaemia.
Radiotherapy: LN causing discomfort or obstruction.& for symptomatic splenomegaly Splenectomy : may be , to improve low Bl. Count. due to autoimmune destruction or due to hypersplenism & can relieve massive splenomegally.
* Prognosis: Overall survival is 6y., stageC 2-3y, ,50% die from infection.
Rarely → Aggressive high grade Lymphoma(Richters syndrome).
Prolymphocytic Leukemia
* Variant of CLL, mainly in Male ˃ 60y. 25% T cell variety* Massive splenomegaly, + Little Lymphadenopathy.
* WBC often ˃ 400 x 10 9/ L (Characteristic cell is large lymphocyte with prominent nucleolus.
* Treat. : Is generally unsuccessful & prognosis very poor.
Leukapharesis, Splenectomy, & chemo. May be tried.
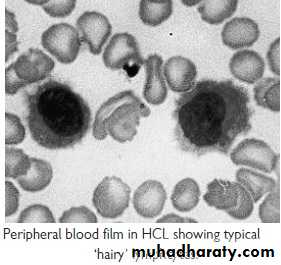
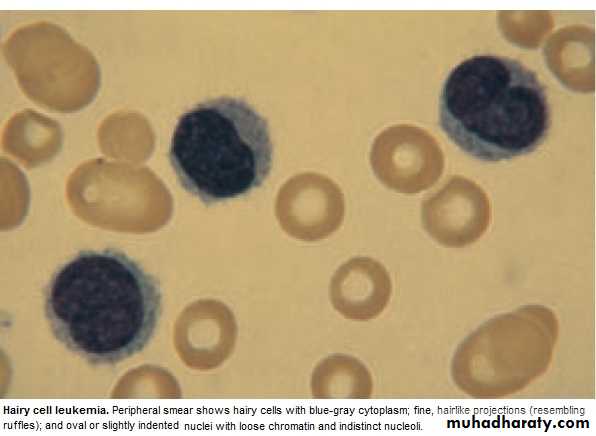
Hairy cell Leukemia :
* Rare,chronic lymphoproliferative B cell disorder, M 6:F 1 , median age 50y.* General ill health, & recurrent infection.
* Splenomegaly 90%, LN unusual.
* Lab.: Severe Neutropenia, Monocytosis,& characteristic HAIRY CELL in Bl. & B.M..
→ CD25 & CD103.
* Treat.: Cladribine & Deoxycoformycin.
Summary
Acute leukemias: ALL & AMLFailure of maturation.
Lab: > 20% Blast.in BM.
R: Supportive or aggressive
CML:↑profil. Of all haemopo.cells.
90% +ve ph chr.
3 phases: Acute, Accelerated, Blast.
R:Imatinib
CLL: B Lymphocytes fail to transform & produce Ab.
3 stages.
R:Fludarabine
Objectives
LymphomasHodgkin
Non- Hodgkin
Paraproteinaemias
Lymphomas
* Neoplasm arise from lymphoid tissues. majority are of B cell origin. diagnosed from pathological changes on biopsy as Hodgkin or Non-Hodgkin lymphoma.
* Non-Hodgkin lymphomas are classified as:
1. High - grade: rapidly dividing, present for Wks.before diagnosis, life threatening.
2. Low - grade: slowly dividing, present for Mns.before diagnosis, indolent.
Hodgkin lymphoma (HL)
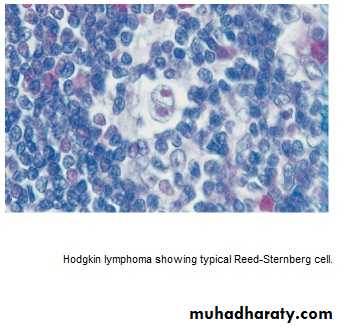
* Histological hallmark is Reed –Sternberg cells.
* 4/ 100 000/y, slight M. excess, median age 31y
Aetiology unknown, ↑in educated background & small families, X3 ↑ likely with
PMH of inf. Mononucleosis ,(no association with EBV).
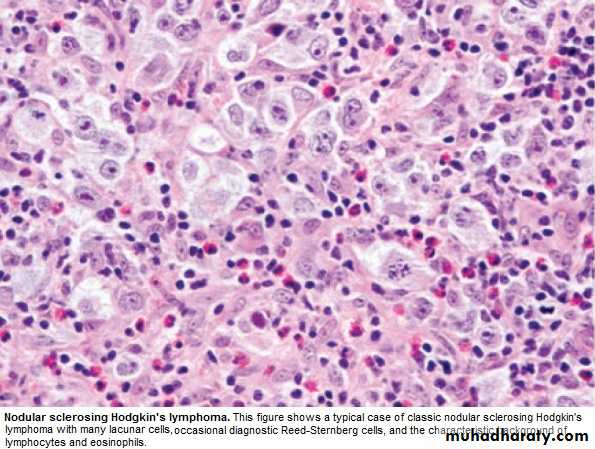
* Pathological classification:
1.Nodular lymphocyte predominant,(5%),slow growing, localized, rarely fatal.
2.Classical HL:
a. Nodular sclerosing (70%), ↑in young F.
b.Mixed Cellularity (20%), ↑elderly.
• c. lymphocyte-rich: (5%) ↑M.
d.Lymphocyte –depleted : (rare)
Lecture (8)
Clinically :
* PAINLESS,RUBBERY LN-pathy, ↑ neck,& supraclavicular. Young with Nod. Sclerosis may have large mediastinal masses (asymptomatic or dry cough & SOB).Isolated subdiaphragmatic nodes ˂ 10% at diagnosis.
* Hepatosplenomegaly may be present but not always indicate dis. In them.
* Extra nodal spread to bone brain or skin --- rare.
Clinical Stages: (Ann Arbor)
1.Stage I : Single LN region(I)or extra lymphatic site(IE).
2.Stage II: 2 or more LN regions(II),or an extralymph.site & LN regions on the same side of (above or below) the diaphragm(IIE).
3. Stage III: LN regions on both sides of the diaph.with(IIIE) or without (III) localized
extra lymph. Involv. Or involv. Of spleen(IIIs) or both (IIISE).
4. Stage IV: Diffuse involv. Of one or more extralymph. Tissue e.g. liver or BM.
Note: 1. Each stage is subclassified into :
A. No systemic symptoms.
B. Systemic symptoms ( Weight loss, drenching sweat)
2. Lymphatic structure = LN, Spleen, Thymus, Waldeyers ring, Appendix &
peyers patches.
Investigations:
* FBC: may be Normal. Anemia or Lymphopenia :poor prognostic F.. ± ↑ E , N & ESR.* Renal f. tests: (to insure normal function before treat.).
* Liver f. tests: Abnormal may reflect hepatic infilt., LN in porta hapatis → obs.j.
* LDH: ↑ = Poor prognostic F.
* CXR: Medast mass ?
* CT: chest, abd., pelvis → staging. Bulky dis.= ˃ 1o cm single node mass(poor prog.F) * LN biopsy:surgical or percut. Needle biopsy.
Management:
* Historically: stage IA & IIA : Local radiotherapy(Radio.T)
*.Recently: Early-stage dis.has better outcome if chemotherapy(Chemo.T) included.
Majority are now treated with Chemo,T.+ Radio.T.
ABVD regimen (doxorubicin,vinblastine,bleomycin & dacarbazine).
Early –stage: 4 courses ABVD,followed by radio.T. to involved LN.
Response by CT & positron emission tomography(PET).
S.E.of ABVD: doxorubicin → cardiac,bleomycin → pulmonary.
↓ MDS /AML & infertility.
Advanced stage: Chemo.T. alone. 6-8 cycles ABVD., Therapy resistant → BMT.
Prognosis:
Early- stage: 90% CR, majority cured.Advanced –stage: 45-70% cured.
Relapse within a year → good salvage rate with autologous BMT.
Relapse after 1y → Chemo.T
Non- Hodgkin lymphoma ( NHL )
* Monoclonal prolif. Of lymphoid cells of B cell (70%) or T cell (30%) origion.* 12 new cases / 100 000/y. slight ↑ M ,median age 65-70y.
Aetiology:
* Late manifest. Of HIV.
* EBV,HTLV.(Certain type of lymphoma).
* H pylori –Gastric lymphoma.
*Chromosomal lesion: t(14:18) –Follicular lymphoma.
*Immunosuppressed pts. after organ transplant, congen. immunodef states.
Clinically: The most important factor is grade:
* High grade: High proliferation rate, rapidly producing symptoms, fatal if untreated, potentially curable.
* Low grade: Low prolif. rate, may be asymptomatic for many Ms before presentation.
indolent course, not curable.
Other forms include : mantle cell L. &MALT lymphoma (less common).
Clinically:
* Often widely spread at presentation, including extra nodal sites.* LN-pathy + systemic upset: Weight loss, sweats, fever & itching. ± H.splenomegally.
* Extra nodal involves BM, gut, thyroid, lung, skin, testis, brain & bone.
BM involv. is more common in low grade (50-60%) than high grade (10%).
Compression syndrome: gut obst., Ascitis, SVC obst. & SC compression.
* Staging is same as HL, but NHL is more likely to be stage III & IV at presentation.
Investigations: as in HL + the followings:
1.Routine BM aspirate & trephine biopsy.
2.Immunophenotyping of surface antigens to distinguish T & B cell tumors.
3. Igs: some lymphomas are associated with IgG or IgM paraproteins( serve as
markers for treatment response.
4. Uric acid ↑ with treat..
5. HIV.
Management:
Low grade lymphoma:
* Asymptomatic: may not require treat.
Indications: marked systemic symptoms, LN causing discomfort or disfigurement.
* Options:
1. Radiotherapy: localized stage I (rare)
2. Chemotherapy: Chlorambucil (oral), well tolerated but not curative.
More intensive iv chemo. In young → better quality of life
but no survival benefit,
3.Monoclonal Ab. Therapy: target surface Ag.on tumor cells.→ cell apoptosis.
Rituximab(R)(anti-CD20) → durable response in 60%,when given alone.
Ist line therapy = R-CVP ( Cyclophosphamide, Vincristine, Prednisolone)
4.Relapse: ↑ dose Chemo. + BMT (Study is going on).
High grade lymphoma: needs treat. At initial presentation.
1. Chemotherapy: > 90%: CHOP(Cyclophosphamide, Doxorubicin,Vincristine,Predn)
2. Radiotherapy: Few stage I, residual bulky dis. After chemo, compression(e.g. SC.)
3.Monoclonal Ab. Therapy: R-CHOP → ↑ complete response rate & survival.
4.Autologous BMT: relapsed chemosensitive pt.
Prognosis:
Low grade: Indolent, remitting & relapsing,median survival- 10y.High grade: 80% respond initially to treat. But only 35% have dis.-free survival at 5y.
Paraproteinaemia:
Gammopathy: Overproduction of 1 or more Ig.( detected by electrophoresis).Polyclonal: Causes: Acute or chr. Inflamation e.g.Infections, Sarcoidosis, Autoimmune disorder, Malignancy.
Monoclonal:(=M Protein) occurs with N or ↓ other I gs.
Myeloma, Lymphoma, Amyloidosis, CTD e.g. Rh. arthritis ,HIV, Solid
tumors or no underlying dis. gammapathy of uncertain origion (MGUS):
Common condition with increasing age.
Paraprotein is present in Bl. but with no other features of MM, WM,Lymphoma,
or related dis.
Clinical: Usually asymptomatic, normal FBC & Biochemistry. Ig : mild ↑with
no immune paresis. BM → Plasma cells < 10% of nucleated cells.
Prognosis: After 20y → 25% MM or related disorder.
Annual monitoring.
Waldenstrom macroglobulinaemia
Low- grade lymphoplasmacytoid lymphoma associated with IgM paraprotein,causing clinical features of hyperviscocity syndrome. Rare, ↑ elderly, ↑ M.
Clinically: Nose bleed, bruising,confusion & visual disturbance.
May present with An.,systemic symptoms, splenomegaly,or LN-pathy.
Lab.: ↑ IgM + ↑ plasma viscocity.
BM: Infiltration with lymphoid cells & prominent mast cells.
Manag.:
Severe hyperviscocity & An. → plasmapheresis.
Chlorambucil: effective but slow.
Fludarabine: more effective.
Rituximab: also effective
Summary
Hodgkin Lymphoma: Histological ly: Reed –sternberg cells.High & Low grades.
Unknown etiology.
Painless LN ± Hepatosplenomegally.
R : ABVD.
Prognosis: Majority cured.
Non-Hodgkin Lymphoma:
High & Low grades.
High grade is potentially curable.
Often wide spread at presentation + systemic symptoms.
R : Low grade: R-CVP.
High grade:R-CHOP.
Objectives:
Multiple Myeloma.Myeloproliferative diseases:
Myelofibrosis.
Essential Thrombocythemia.
PRV\
Infectious mononucleosis.
Multiple Myeloma (MM) Lecture 9
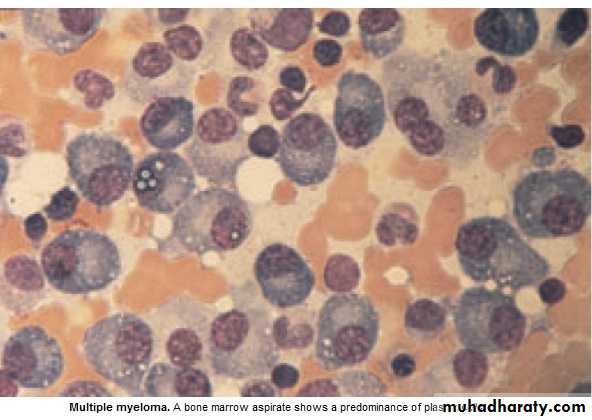
* Malignant prolif. Of plasma cells.
Normally: Plasma cell produce polyclonal Ig = variety of heavy chains are produced,
& each may be of kappa or lambda light chain type.
Myeloma: Plasma cells produce Ig of a single heavy & light chain (monoclonal prot-
ein = paraprotein), in some cases only light chain is produced →appear
in urine (Bence Jones proteinuria).
Classification:
Type of paraprotein %
IgG 55
IgA 21
Light chain only 22
Others(D,E, non-secretory) 2
*Majority of malignant plasma cells are present in BM ,small No. in circulation.
*Malignant plasma cells produce cytokines → stimulate osteoclast →bone reabso-
rption. → lytic lesions → pain, fracture, & ↑ Ca++.
*BM involv. → An. or pancytopenia.
Clinically:
4/100 000/y, M:F 2:I,Median age 60-70y.PATHOLOGY EFFECTS SYMPTOMS
Marrow involv. Bone erosion by osteoclast Pain
path. Fracture Severe local pain
↑Ca++ Lethargy,thirst.
BM failure → An. Tiredness
↑ paraprot.& L chain Renal damage Renal failure
↑ viscocity Blurred vission, headach
Amyloidosis Nephrotic syndrome
↓ Normal plasma cells ↓ immunity ↑ infection(UTI,Resp.)
Diagnosis: Requires 2 of the followings:
1. BM malignant plasma cells >20% (BM aspiration)
2. S & or urinary paraprotein.(Bl & urine protein electrophoresis)
3. Skeletal lytic lesions.(X-rays/skeletal survey)
other investigations:
1. FBC: (Degree of BM failure),↑ ESR (not specific), urea & electrolytes, creatinine, uric ac., S.Ca++ & albumin.
coagulaton screen?, B2 microglobulin, MRI( SC compr.).
2.Bl. & urine immunoelectrophoresis(type of paraprotein).
3.Quantification of paraprotein,& other Igs( ↓ = immune paresis).
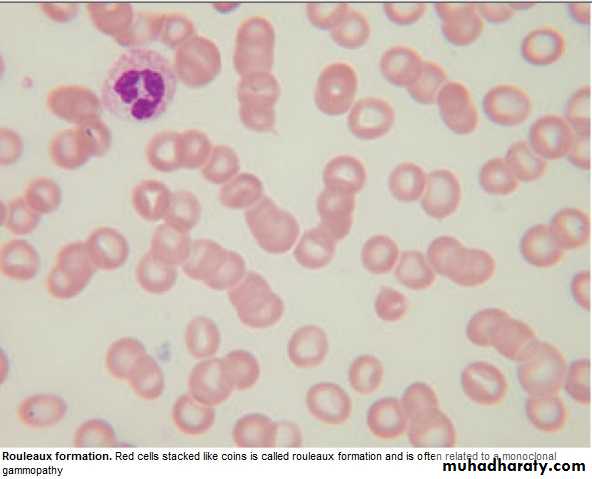
Management:
Asymptomatic: treat. may not be required.Immediate support:
1. High fluid intake to treat renal impairment & hyper Ca++.
2. Analgesia for bone pain.
3. Biphosphanates for hyperCa++(it also reduce bone pain & risk of fracture, may cause apoptosis of malignant plasma cells)
4.Allopurinol:prevent urate nephropathy.
5. Plasmapheresis for hyperviscocity.
Chemotherapy:
1. Old pts.: Ist line chemotherapy: Thalidomide + Melphalan + Prednisolone →
median survival 51 Ms. 2. Young Pts.: Ist line chemo. until max. response(plateau phase),then autologous
BMT (prolong survival but no cure).
3. Relapse: Bortezomib.
Radiotherapy:
For localized pain not responding to analgesia, pathological fracture & for emergency treat. Of SC compr. Complicating extradural plasmacytoma.
Prognosis: Poor prognostic features: ↑ B2-microglobulin, ↓ albumin, ↓Hb or ↑Ca++ at presentation. 5% survive 5y.
Myeloproliferative disorders:
Group of diseases characterized by clonal prolif. Of marrow erythroid precursors(PRV),
Megakaryocytes( Essential thrombocythaemia & Myelofibrosis) or myeloid cell CML), some having overlapping features,& often progression from one to another e.g. PRV to myelofibrosis.
MYELOFIBROSIS:
BM is initially hypercellular, with excess abnormal megakaryocytes which release
growth factors e.g. platelet-derived growth factor, to the marrow microenvironment
→ reactive prolif. of fibroblasts →marrow fibrosis.
Clinically: Age ↑ ˃ 50y, Lassitude, wt. loss, night sweat. SPLEEN MASSIVELY ENLARGED (extramedullary haematopoiesis).
Lab.: 1.FBC: Leukoerythroblastic An. ↑ reticulocyte count, tear drop RBC. WBC: ↓ - ↑ .
Platelets: ↑ , N, ↓, Giant form may be seen. 2. ↑uric ac., folate def. is common.
3. BM aspirate: difficult, Biopsy: Excess megakaryocytes,↑ reticulin & fibrous T. replac.
4. JAK -2 mutation 50%.
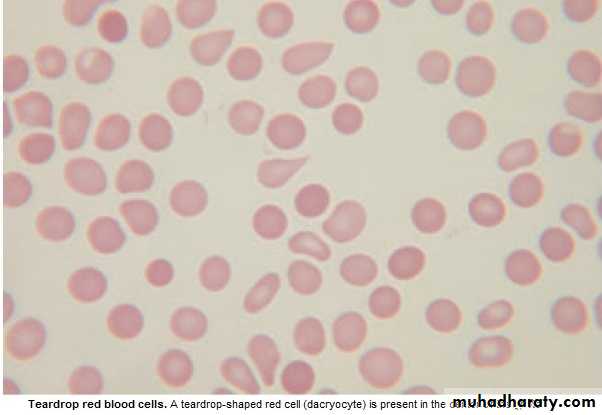
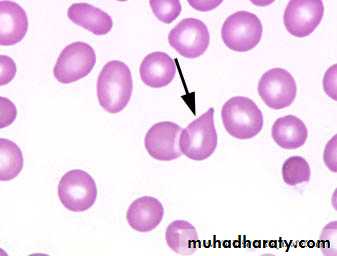
Management & prognosis:
Median survival: 4y.( range 1-20y).Treat.: Control symptoms:
RBC transf. for An.
Folic ac. To prevent def.
Hydroxycarbamide to control spleen size, WBC count, systemic symptoms.
Splenectomy: grossly enlarged, or pancytopenia(hypersplenism)
BMT: may be considered for young pt.
Essential thrombocythaemia;
Malignant prolif. Of megakaryocytes result in raised level of circulating platelets,that are often dysfunctional.
Reactive causes of ↑plat. must be excluded.
Clinically: Median age: 60y, may be asymptomatic with ↑ plat. Count.
Vascular occlusion or Bleeding.
Small % → Acute leukemia or Myelofibrosis.
Lab.: ↑ plat.,JAK-2mutation 50%
Treat.:
* ↓ risk pt.: ( age < 40y, plat.c. < 1000 x 10 9/L & no bleeding or thrombosis) →
may not require treat.
* Plat, c. > 1000 x 10 9/L, with symptoms, or with risk factors for thrombosis(DM,
HTN),→ Treat. to control plat.c../ e.g. Hydroxycarbamide or Anagrelide(inh. Of
megakaryocytes maturation). Radioactive phosphorus( P32) for old age.
* Aspirin for all pts. to reduce risk of thrombosis.
Infectious mononucleosis (IM)
*Acute viral illness, most often caused by EBV(CMV, HIV-1 & Toxoplasmosis may
cause similar clinical syndrome).acquired from asymptomatic excreters via saliva
by droplet infection or kissing.
Clinically: I.P.:Prolonged, undetermined.
Fever, Headache,& malaise,followed by severe pharyngitis (± tonsillar exudate, Ant. & Post. cervical LN-pathy), palatal petechiae, periorbital oedema,splenomegaly, inguinal & axillary LN. & macular, petechial,or erythema multiforme rashes may
occure. Fever resolved in 2 wks & fatigue in another 2 wks.
Death is rare : resp.obst., splenic rupture, thrombocytopenia, or encephalitis.
Investigations: 1. FBC: Atyical Lymphocytes in peripheral Bl.
2. Paul-Bunnel or Monospot test: detect heterophil Ab.present during acute illness
& convalescence (to be repeated if initially –ve).
3.Specific EBV serology (immunofluorescence) can be used to confirm the diagnosis
Complications:
Common Uncommon RareSevere pharyngeal Odema Neurological Ruptured spleen
Antibiotic-induced rash Cranial nerve palsies Resp. obstruction
(80-90% with Ampicillin) Polyneuritis Agranulocytosis
Prolonged post-viral fatigue Transverse myelitis Lymphoprplif. Dis.
Hepatitis(80%) Meningoencephalitis
Jaundice( <10%) Hematological Hemolytic An. Thrombocytopenia Renal Abnormal GUE Interstitial nephritis Cardiac
Myocarditis, abnormal ECG
Pericarditis
Management:
1.Symptomatic:2.If throat culture revealed B-haemolytic streptococci → penicillin.(avoid
ampicillin
& amoxicillin → macular rash .
3.Severe pharyng. Odema → prednisolone 30 mg 5d.
4.Antiviral: not effective.
5.Avoid contact sports (splenic rupture) until splenomegay has completely
resolved .
Summary
MM:IgG is the most common type.
Diagnosis 1. > 20% plasma cells in BM.
2.Serum or Urine paraprotein.
3.Skeletal lytic lesions.
R: Thalidomide + Melphalan+ Prednisolone
Myelofibrosis:
Massive splenomegally.
R: Symptomatic ± Hydroxycarbamide.
Splenectomy, BMT.
ET: ↑plat. clinically: vascular occlusion or bleeding.
R: Aspirin + Hydroxycarbamide when indicated.
Infectious mononucleosis: EBV, Atypical L, Paul-Bunnel,symptom.R.