
0
Mustafa Hatim Kadhim
Baghdad University
Al-kindy college of medicine
Third Stage
2013 - 2014

1
List of contents
Lecture
number
Lecture name
Doctor name
Page
number
Unit 1 - Good medical practice
2 - 5
1
Bedside Medical Teaching
دكتور
علي الساعدي
3 - 5
Unit 2 - Molecular and genetic factors in disease
6 - 18
1
Introduction
دكتورة
اخالص
7 - 8
2
Classification of Genetic disorders
9 - 12
3
Clinical Presentation of Diseases with molecular
defect - General principles of diagnosis
13 - 15
4
Gene Therapy
16 - 18
Unit 3 - Immunological factors in disease
19 - 35
1
Functional Anatomy & Physiology of the immune
system
دكتور
ة
بتول
20 - 23
2
Immune deficiency
24 - 27
3
The Inflammatory Response
28 - 30
4
Autoimmune disease
31 - 32
5
Allergy - Transplantation and graft rejection
33 - 35
Unit 4 - Infectious disease
36 - 51
1+2+3
Protozoal Infections
دكتور
حيدر
37 - 44
4+5+6+7
Infections caused by Helminthes
45 - 51
Unit 5 - Pain
52 - 65
1
Introduction
دكتور
زيدان
53 - 54
2
Headache
55 - 57
3
Chest pain
58 - 61
4
Abdominal pain
62 - 65
Unit 6 – Common diseases
66 - 81
1
Cough & Hemoptysis
دكتور
زيدان
67 - 69
2
Breathlessness (Dyspnea) & Cyanosis
70 - 72
3
Dysphagia
73 - 74
4
Hematemesis, Melena & Diarrhea
75 - 76
5
Constipation & Jaundice
77 - 78
6
Oedema & Ascites
79 - 81
Unit 7 - Clinical biochemistry and metabolism
82 - 92
1+2+3
دكتور
علي الساعدي
83 - 92

2

Unit 1 - Good medical practice
3
Bedside Medical Teaching
To study medicine at bedside of your patient
Talk with a patient
Take the history from the patient
Examine the patient
Formulate your findings into differential diagnosis
Rank these in order of probabilities
Use investigations to support or refute your differential
diagnosis
Listen to your patient, he is telling you the diagnosis.
The doctor may also learn more about the illness from the
way the patient tells the story than from the story itself.
Dr. James B. Herriock (1861-1954).
The most vital part of a doctor is his interest in humanity.
Always remember that your patient is a human being ,he
is hopeful but fearful.
(Harrison-Principles of internal Medicine).
The first and most important step for a successful doctor
is to educate the patients so that they can decide what is
the best course and procedure for themselves
Do you really need this?
(Half of what you are taught as medical student will in ten
years have been shown to be wrong and the trouble is
none of your teachers know which half)
Dr. Sydney Burwell
Impact of medical information research showed changes
in patient management choice of medication 45% and
diagnosis 29%
High cost of not finding information
New beginning
New dreams
New memories
Main defect
Low awareness among population and patients
Observer error
You see only what you look for
You recognize only what you know
Merril C. Sosman,Bosten
Close friends and relatives provide a useful third-party
view of the patients well being
The duties of a doctor registered with the UK
General Medical Council
Patients must be able to trust doctors with their lives and
health. To justify that trust you must show respect for
human life and you must:
Make the care of your patient your first concern
Protect and promote the health of patients and the public
Provide a good standard of practice and care
Keep your professional knowledge and skills up to date
Recognize and work within the limits of your competence
Work with colleagues in the ways that best serve patients'
interests
Treat patients as individuals and respect their dignity
Treat patients politely and considerately
Respect patients' right to confidentiality
Work in partnership with patients
Listen to patients & respond to their concerns & preferences
Give patients the information they want or need in a way
they can understand
Respect patients' right to reach decisions with you about
their treatment and care
Support patients in caring for themselves to improve and
maintain their health
Be honest and open, and act with integrity
Act without delay if you have good reason to believe that
you or a colleague may be putting patients at risk
Never discriminate unfairly against patients or colleagues
Never abuse your patients' trust in you or the public's trust
in the profession.
You are personally accountable for your professional
practice & must always be prepared to justify your
decisions & actions
Change management
The only constant in medicine is change
Medicine is an ever changing subject
Opinion base evidence base
MOH

Unit 1 - Good medical practice
4
Facts and figures
No percentage
Should believe in change
5 star doctor: tomorrow's doctor...good doctor
WHO goal
Leader
Communicator
Team worker
Policy maker
Care provider
Multidiscipline sheet
Paramedics
Nurse
Resident
Senior House Officer
Clinical Pharmacist
Dietician
Specialist senior / Senior Consultant
administrator / Statistician
Evidence base data
Young platform doctors
Thinking / Attitude
Opinions
Sharing actively in duties
Handworkers / breadwinners
Future pioneers
Rewards to youngest researcher
Chance to postgraduate candidate to hold & present talks
Residents are the future:
Medicine is a MISSION not a business
The most vital part of a doctor is HUMANITY
Always remember that your patient is a HUMAN BEING
He is FEARFUL BUT HOPEFUL
Seniors
Wisdom / Experience
Father figure
supervisor
Policy and decision maker
Law Maker
Insanity repeat things over & over & you think you are
doing something
New Creativity repeat things to have something new
Innovation create something new
Change syllabus According to
Community demands
Stakeholders
Doctor patient relationship
Third party
Practicalities / Block System
flooding technique
Knowledge
Skills
Attitude
Attitude Problem
Ego / Super Ego
Cooperation coordination collaboration

Unit 1 - Good medical practice
5
Education is the most powerful weapon which you can
use to change the world. Nilson Mandella
The biggest enemy of health in the developing world is
poverty. Koffi Anaan (The secretory general UN
&Noble price of peace 2001)
Medicine is a social science and politics is nothing but
medicine on a larger scale
RUDOLF VIRCHOW (1821-1902)
While medicine is getting increasingly molecular, don't
lose the human side of it
االستاذ الدكتور محمد علي خليل المدامغة

6

Unit 2 - Molecular and genetic factors in disease
7
Lecture 1 - Introduction
Molecular medicine
Is a broad field, where physical, chemical, biological and
medical techniques are used to:
Describe molecular structures and mechanisms
Identify molecular and genetic errors of disease
Develop molecular interventions to correct them. The
molecular medicine perspective emphasizes cellular and
molecular phenomena and interventions rather than the
previous conceptual and observational focus on patients
and their organs
Classification of Human diseases
1) Those that is genetically determined.
2) Those that is almost entirely environmentally determined.
3) And those to which both nature and nurture contribute.
About 1% of all newborn infants possess a gross
chromosomal abnormality.
Approximately 5% of individuals under age 25 develop a
serious disease with a significant genetic component
The human genome
The gene is the most important unit of genetics.
The estimated total number of genes is about 30000-
35000, the gene has an average 1400 base pairs,only 1.5%
of the genome reprsents primary coding sequence
There are 3×10
9
(3000 megabase) base pairs of deoxy-
ribonucleic acid (DNA) present In the human genome.
DNA forms a double stranded helical structure with only
two type of base pairing are possible G-C and A-T, the
double stranded DNA unit of two nucleotides is referred
as a (BASE PAIR (bp))
The double stranded DNA helix is coiled around
chromosomal protein called histone to form nucleosomes
then these also adopt a coiled structure to form a
chromatin fibre which on more coiling forms the
chromomosome
DNA contains all of the genetic information required for
the development of cells into tissue & organs
Human cells contain 46 chromosomes:
22 pairs of autosomes and 1 pair of sex chromosome
One chromosome of each pair is derived from each
parent; these 46 chromosomes are the diploid number
seen in somatic cells.
Only germ cells (sperm &ova) have the haploid number
of 23 chromosome (22 autosomes and either an X or Y)
The genetic code is a 3 nucleotides unit which specify
certain aminoacid to be inserted into protein ,only a very
small fraction of the human genome codes for protein;
a Locus refers to any area of the genome ,
not all the DNA code for protein
sequences within the gene include coding
regions(exons),noncoding regions(introns)and regulatory
sequences
The conversion of DNA sequence to protein is mediated
by RNA
Most of the natural variation in DNA sequence occurs in
the noncoding region and have no effect on development
and function ,
variations that occur in the coding region leads to change
in protein sequence and possibly the function
If DNA variation results in sufficient impairment of
protein function to bring a harmful effect then A
GENETIC DISEASE may result.
When the genetic abnormality is mild, the genetic disease
can be recognized, but sometimes the genetic abnormality
may impair more vital process to such an extent that
embryogenesis cannot continue.
X inactivation
Is a special property of the X chromosome in the female
one of the two X chromosome in a cell is inactive, so in a
similar manner to male ,females only express one copy of
genes on the X chromosomes and this process of
inactivation is random ,
This can have a bearing on the expression of diseases
which are due to mutations in genes on the X
chromosome as either the normal or the mutant gene may
be inactivated.
Genotype: is the genetic makeup of an individual (the
sequences of their gene)
Phenotype: is any aspect of structure or development of
an individual (is the "outward, physical manifestation" of
the organism)
Genomic imprinting
• It means that the effect of a gene depends on whether its
inherited from the mother or father
• If an imprinted gene carries a mutation then the
manifestation of the disease will vary according to which
parent transmitted the mutation.
• For example a certain region on chromosome 15 contain
several genes in which only the paternal or maternal allel

Unit 2 - Molecular and genetic factors in disease
8
is transcriptionally active so deletion of these gene on the
paternal chromosome cause a syndrome called (prader
willi syndrome) while deletion of maternal chromosome
causes a different syndrome(angelman's syndrome)
Polymorphism
It represents a small change in DNA sequence that does
not result in overt diseases this can happen if:
The change occurs in the noncoding DNA.
Do not alter the amino acid inserted in a given protein.
Result in an amino acid which is able to perform the same
function as the original.
We all share genome sequence that are 99.9% identical,
the remaining 0.1% is therefore responsible for all genetic
diversity between individuals.
Genetic factors in common diseases
Susceptibility to many common diseases is influenced by
genetic factors, this is recognized by an increased
incidence of the diseases in first degree relatives of
affected individuals but not in a pattern typical of
classical single gene disorders
Asthma,hypertention,diabetes,atopy , and ischemic heart
diseases and many infectious diseases show this pattern
Genes that act together with environmental influences
giving rise to this susceptibility are of interest
Penetrance and expression
Individuals who inherit a specific disease mutation do not
show an identical phenotype since they may not share the
other genetic or environmental factors that predispose or
unmask the full effect of the mutation.
Penetrance is the proportion of individuals who develop
the diseases phenotype.
Fully penetrant mutation: if all individual who inheret it
Develop the associated disease phenotype.
If additional environmental factors are needed the gene
may show late onset pentrance
Non –pentrant mutation if the individual is not exposed
to sufficient additional factors
Disease Expression describes the degree to which the
severity of the disease phenotype varies.
Genetic counseling
Genetic counseling is providing information about the
medical and family implications of a specific disease.
Aims of Genetic counseling is to:
Help individuals make decisions about planning a family
Taking part in screening program
Accepting prophylactic therapies.
Specific problems encountered in genetic counseling
include:
Accurate assessment of genetic risk
Identification of children at risk of genetic disorders
The increase in genetic risks associated with
consanguinity
Non-paternity as an ncidental finding in DNA diagnostic tests
Indication of genetic counseling
One or more birth defects
A genetic disorder or chromosome abnormality
Intellectual Development Disorder or developmental delay
Neuromuscular abnormalities
Unexplained metabolic problems
Congenital or familial hearing loss or blindness
Abnormal growth patterns
Abnormal sexual development
Prenatal exposure to drugs or medications
Cancer

Unit 2 - Molecular and genetic factors in disease
9
Lecture 2 – Classification of Genetic
disorders

Unit 2 - Molecular and genetic factors in disease
10

Unit 2 - Molecular and genetic factors in disease
11

Unit 2 - Molecular and genetic factors in disease
12

Unit 2 - Molecular and genetic factors in disease
13
Lecture 3 –
Clinical Presentation of Diseases with
molecular defect
1. Inborn errors of metabolism
Inborn errors of metabolism (IEM) are caused by
mutations that disrupt the normal function of a
biochemical pathway. Most IEM are due to autosomal or
X-linked recessive loss-of-function mutations in genes
encoding specific enzymes or enzymatic co-factors.
Knowledge of the biochemical pathway involved means
that specific blocks have predictable consequences,
including deficiency
of the end product and build-up of intermediary
compounds. Many hundreds of different IEM have been
identified and. Most IEM are restricted to paediatric
practice; however, a growing number may now present
during adult life and some of these are discussed below:
Intoxicating IEM
A subgroup of IEM, termed ‘intoxicating IEM’, can present
as a sudden deterioration in a previously well individual.
Such deteriorations are usually precipitated by some form
of stress, such as infection, pregnancy, exercise or changes
in diet. The intoxication is due to the build-up of
accumulation of intermediary compounds, which will vary
according to the pathway involved. For example, in urea
cycle disorders ammonia is the toxic substance
The intoxication is often associated with derangement of
the acid–base balance and, if not recognised and
treated,will often proceed to multi-organ failure, coma
and death.
The diagnosis of these disorders requires specialist
biochemical analysis of blood and/or urine.
Treatment relies on removal of the toxic substance using
haemodialysis or chemical conjugation, and prevention of
further accumulation by restricting intake of the precursors:
as total protein restriction in urea cycle disorders
Mitochondrial disorders
Disorders of energy production are the most common type
of IEM presenting in adult life
The tissues that are most commonly affected in this group
of disorders are those with the highest metabolic energy
requirements, such as muscle, heart, retina and brain.
Therapy in this group of disorders is based on giving
antioxidants as vitamin C and co-factors that can improve
the function of the respiratory chain.
Storage disorders
Storage disorders are most commonly caused by loss-of
function mutations affecting enzymes involved in
lysosomal degradation pathways.
The clinical consequences depend on the specific enzyme
involved. For example:
Niemann–Pick disease type C is caused by autosomal
recessive loss-of function mutations in either the NPC1 or
NPC2 gene.
These results in hepatosplenomegaly, dysphagia, loss of
speech, early dementia, spasticity, An increasing number of
storage disorders are treatable with enzyme replacement
therapy, making awareness and diagnosis more important.
2. Neurological disorders:
Progressive neurological deterioration is one of the most
common presentations.These diseases are mostly
autosomal dominant , examples would be early-onset
familial forms of dementia, Parkinson’s disease ,
Huntington disease The triplet repeat disorders cause an
interesting group of syndromes and have specific features
Huntington disease:
Huntington disease (HD) is triplet repeat disorders. This
condition can present with
a movement disorder or
weight loss or
psychiatric symptoms (depression, psychosis, dementia)
Or with a combination of all three.
The disease is the result of a [CAG] triplet repeat
expansion mutation in the HD gene on chromosome 4.
Since CAG is a codon for glutamine, the mutation
probably leads to gain of function, as deletions of the gene
do not cause HD, expansion of the repeat to above the
normal range results in neurological disease.
The severity of disease and age at onset are related to the
repeat length. In HD, atrophy of the caudate nuclei is
obvious on magnetic resonance imaging (MRI) of the
brain, and in later stages cerebral atrophy is also apparent.
There is currently no therapy that will alter the
progression of the disease, which will often be the cause
of the patient’s death.
Within families there is a tendency for disease severity to
increase and age at onset to fall due to further expansion
of the repeat, a phenomenon known as anticipation. The
mutation is more likely to expand through the male germ
line than through female.

Unit 2 - Molecular and genetic factors in disease
14
3. Connective tissue disorders
Mutations in different types of collagen, fibrillin &elastin.
Make up the majority of connective tissue disorders.
The clinical features of these disorders vary, depending on
the structural function and tissue distribution of the
protein which is mutated.
For example, autosomal dominant loss-of-function
mutations in the gene encoding elastin cause either
supravalvular aortic stenosis, The most commonly
involved systems are:
skin (increased or decreased elasticity, poor wound
healing)
eyes (myopia, lens dislocation)
blood vessels (vascular fragility)
bones (osteoporosis, skeletal dysplasia)
joints (hypermobility, dislocation, arthropathy
)
4. Learning disability, dysmorphism &
malformations
Congenital cognitive impairment (also called mental
handicap or learning disability) affects about 3% of the
population.
There are important ‘environmental & Genetic’ causes of
cognitive impairment, including:
teratogen exposure during pregnancy
(alcohol,anticonvulsants)
Congenital infections (cytomegalovirus, rubella,
toxoplasmosis, syphilis)
premature delivery (intraventricular haemorrhage)
Birth injury (hypoxic encephalopathy).
Genetic disorders that contribute to the etiology of
cognitive impairment are (Chromosome disorders &
dysmorphic syndromes)
Chromosome disorders
Any significant gain or loss of autosomal chromosomal
material (aneuploidy) usually results in learning disability
and other phenotypic abnormalities, Down’s syndrome is
the best known of these disorders, The DNA analysis can
identify causative structural chromosome abnormalities in
10–25% of cases of significant learning disability.
Dysmorphic syndromes
Almost all dysmorphic syndromes are characterized by
the occurrence of cognitive impairment, malformations
and a distinctive facial appearance associated with various
other clinical features.
Making the correct diagnosis is important, as it has
implications on immediate patient management, detection
of future complications and assessment of recurrence risks
in the family.
Clinical examination remains the mainstay of diagnosis
and the patient often needs to be evaluated by a clinician
who specializes in the diagnosis of these syndromes.
The differential diagnosis in dysmorphic syndromes is
often very wide and this has resulted in computer aided
diagnosis becoming an established clinical tool.
The clinical diagnosis may then be confirmed by specific
genetic investigations, as the genetic basis of a wide range
of dysmorphic syndromes has been identified.
5. Familial cancer syndromes
Most cancers are not inherited but occur as the result of an
accumulation of somatic mutation. However some
families are prone to one or more specific types of cancer
affected individuals tend to present with:
Tumors at an early age and are more likely to have
multiple primary foci of carcinogenesis.
Example
Hereditary non-polyposis colorectal cancer
Hereditary non-polyposis colorectal cancer (HNPCC) is
an autosomal dominant disorder with mutations can occur
in several different genes encoding proteins involved in
DNA mismatch repair that presents with early onset
familial colon cancer, particularly affecting the proximal
colon. Other cancers, such as endometrial cancer, are
often observed in affected families.
Familial breast cancer
Familial breast cancer is an autosomal dominant disorder
that is most often due to mutations in genes encoding
either BRCA1 or BRCA2. Both these proteins are involved
in DNA repair. Individuals who carry a BRCA1 or BRCA2
mutation are at high risk of early-onset breast and ovarian
tumours, and require regular screening for both these
conditions. Many affected women will do Prophylactic
bilateral mastectomy and oophorectomy.

Unit 2 - Molecular and genetic factors in disease
15
General principles of diagnosis
Diagnosis can be made by a careful clinical history and
examination and an awareness and knowledge of rare
disease entities. Although DNA-based diagnostic tools are
now widely used, it is important to be aware that not all
diagnostic genetic tests involve analysis of DNA. For
example, a renal ultrasound can detect adult polycystic
kidney disease. By definition, all genetic testing (whether
it is DNA-based or not) has implications both for the
patient and for other members of the family.
1. Clinical history and examination including
constructing a family tree
The family tree—or pedigree—is a three-generation
family history it may reveal important genetic information
of relevance to the presenting complaint, particularly
relating to cancer. A pedigree must include details
from both sides of the family
any history of pregnancy loss or infant death,
consanguinity,
Details of all medical conditions in family members.
dates of birth and
Age at death.
2. Non DNA-based diagnostic tools:
It may sometimes be more economical or convenient to
measure enzyme activity rather than sequencing the
coding region of the genes involved.
Haemoglobinopathy as sickle-cell disease can be
diagnosed by haemoglobin electrophoresis
Immune deficiencies as hypogammaglobulinaemia can
be diagnosed by Ig levels, Complement levels
Inborn errors of metabolism e.g. phenylketonuria can
be diagnosed by Enzyme assays, amino acid levels
Endocrine disease e.g. congenital adrenal hyperplasia by
Hormone levels, enzyme assays
Renal disease can be diagnosed by e.g. polycystic kidney
can be diagnosed by Radiology, renal biopsy
3. DNA-based diagnostic tools
Polymerase chain reaction (PCR) and DNA sequencing:
PCR involves amplification of DNA from small
quantities of starting material. It is the most important
technique in DNA diagnostic analysis. Almost any tissue
can be used to extract DNA for PCR analysis, but most
commonly, a sample of peripheral blood is used. The
ability to determine the exact sequence of a fragment of
DNA amplified by PCR is also of critical importance in
DNA diagnostics.
((
لالطالع: To detect the mutant gene, two primers (lengths of a
single stranded DNA made complementary to part of the
gene to be tested that bind to the 3' and 5' ends of the normal
sequence are designed. By using appropriate DNA polymerases
(enzymes that build up DNA strand based on its complementary
strand) and thermal cycling, the DNA between the primers is
greatly amplified, producing millions of copies of the DNA
between the two primer sites. The amplified normal DNA and
patient's DNA are then digested with a restriction enzyme that
cuts the amplified DNA into pieces of known sizes e.g. the
normal DNA yields three fragments (67 base pairs, 37 base
pairs, and 163 base pairs long); by contrast, the patient's DNA
yields only two products, an abnormal fragment that is 200 base
pairs (instead of two pairs of 37 and 163 b.p.) and a normal
fragment that is 67 base pairs long. These DNA fragments can
determind by gel electrophoresis (by which we can separate
DNA bands or pieces according to their molecular weight) and
then visualized after staining with special stain under ultraviolet
light..
Hybridization:
This is a procedure used in the diagnosis of genetic and
other pathologies as well as in the diagnosis of cancer.
It is based on the fact that the two DNA strands are not
identical but complementary.
The test is performed by adding a synthetic, single
stranded DNA sequence (called a probe) [that is made
complementary to a specific region of DNA under study
and is being labeled with a specific dye] to the double
stranded DNA from the patient (after making it single
stranded by a process called denaturation). If the probe
found its complementary region along the patient's DNA,
it'll combine (hybridize) to it and starts emitting a color or
"fluoresce". This emitted color can be detected using a
UV-microscope.
This procedure forms the basis of what is known as
fluorescent in situ hybridization (FISH).
Nevertheless, this procedure cannot detect single point
mutations or even addition / deletion of 2 or more
nucleotide bases.
So, the technique used for detection of such smaller
defects is usually DNA-based; the most representative and
most commonly used one is polymerase chain reaction
(PCR) that revolutionalized the diagnostic ability of
genetic testing. Most new techniques used nowadays are
PCR-based.

Unit 2 - Molecular and genetic factors in disease
16
Lecture 4 - Gene Therapy
Aim of the lecture
After completion of this lec the student must know
1. The concept of Gene therapy
2. Types of Vector
3. Types of gene therapy
4. Concept of personalized Medicine &Pharmacogenomics
Gene therapy is a form of treatment that involves
introducing genetic material into a person’s cells to fight
or prevent disease A gene can be delivered to a cell using
a carrier known as a “vector.” The most common types of
vectors used in gene therapy are viruses. The viruses used
in gene therapy are altered to make them safe, with
introducing a therapeutic gene in the vector which will be
then transferred to the patient.
Although some risks still exist with gene therapy. The
technology has been used with some success.
Gene therapy for had been tried for a number of diseases,
such as severe combined immunodeficiencies,
hemophilia ,Parkinson's disease, cancer and even HIV.
Several approaches to gene therapy are being tested,
including:
Replacing a mutated gene that causes disease with a
healthy copy of the gene
Inactivating, or “knocking out,” a mutated gene that is
functioning improperly
Introducing a new gene into the body to help fight a
disease
In general, a gene cannot be directly inserted into a
person’s cell. It must be delivered to the cell using a
carrier, or vector.
Vector systems can be divided into:
1) Viral vectors
2) Non-viral vectors
The most common type of vectors is viruses that have
been genetically altered to carry normal human DNA .
Viruses have evolved a way of encapsulating and
delivering their genes to human cells in a pathogenic
manner. It has been tried to use this ability by
manipulating the viral genome to remove disease-causing
genes and insert therapeutic genes.
Target cells such as the patient's liver or lung cells are
infected with the vector. The vector then unloads its
genetic material containing the therapeutic human gene
into the target cell. The generation of a functional protein
product from the therapeutic gene restores the target cell
to a normal state.
Gene therapy can be split into two categories:
1)
, which means exterior (where cells are modified
outside the body and then transplanted back in again). In
some gene therapy clinical trials, cells from the patient’s
blood or bone marrow are removed and grown in the
laboratory. The cells are exposed to the virus that is
carrying the desired gene. The virus enters the cells and
inserts the desired gene into the cells’ DNA. The cells grow
in the laboratory and are then returned to the patient by
injection into a vein. This type of gene therapy is called ex
vivo because the cells are treated outside the body.
2)
, where genes are changed in cells still in the body,
This form of gene therapy is called in vivo, because the
gene is transferred to cells inside the patient’s body.
Types of Gene Therapy
All cells in the human body contain genes, making them
potential targets for gene therapy. However, these cells
can be divided into two major categories: somatic
cells (most cells of the body) or cells of
the germline (eggs or sperm). In theory it is possible to
transform either somatic cells or germ cells.
1)
Gene therapy using germ line cells
results in
permanent changes that are passed down to subsequent
generations. The application of germ line gene therapy is
its potential for offering a permanent therapeutic effect for
all who inherit the target gene. Successful germ line
therapies introduce the possibility of eliminating some
diseases from a particular family, and ultimately from the
population, forever. However, this also raises controversy.
Some view this type of therapy as unnatural, Others have
concerns about the technical aspects. They worry that the
genetic change propagated by germ line gene therapy may
be harmful, with the potential for unforeseen negative
effects on future generations.
2)
Somatic cell therapy
is more conservative and safer
approach because it affects only the targeted cells in the
patient, and is not passed on to future generations.
However, the disadvantage of this type of therapy is that
the effects of somatic cell therapy are short-lived. Because
the cells of most tissues ultimately die and are replaced by
new cells, repeated treatments over the course of the
individual's life span are required to maintain the
therapeutic effect. Transporting the gene to the target cells
or tissue is also problematic. Regardless of these
difficulties, however, somatic cell gene therapy is
appropriate and acceptable for many disorders,
including cystic fibrosis, muscular dystrophy, cancer, and
certain infectious diseases, the results of any somatic gene

Unit 2 - Molecular and genetic factors in disease
17
therapy are restricted to the actual patient and are not
passed on to his or her children. All gene therapy to date
on humans has been directed at somatic cells, whereas
germline engineering in humans remains controversial .
Choices of Vectors
The ideal vector should have the following characteristics:
1) An adequate carrying capacity (some genes are large) .
2) To be undetectable by the immune system.
3) To be non-inflammatory.
4) To be safe to the patients.
5) Efficient
6) To have long duration of expression and or the ability
to be safely readministred.
Viral Vectors
Viruses attack their hosts and introduce their genetic
material into the host cell as part of their replication cycle.
This genetic material contains basic 'instructions' of how to
produce more copies of these viruses, The host cell will
carry out these instructions and produce additional copies
of the virus, leading to more and more cells becoming
infected. Some types of viruses insert their genes into the
host's genome. This incorporates the genes of that virus
among the genes of the host cell for the life span of that
cell.Viruses like this could be used as vehicles to carry
'good' genes into a human cell. First, the virus genes that
cause disease must be removed then those genes are
replaced with genes encoding the desired effect
This procedure must be done in such a way that the genes
which allow the virus to insert its genome into its host's
genome are left intact.
Non-Viral Vectors
Non-viral methods present certain advantages over viral
methods, with simple production and low host
immunogenicity. Previously, their disadvantage was low
levels of transfection and expression of
the gene however, recent advances in vector technology
have generate molecules and techniques with transfection
efficiencies similar to those of viruses.example: .Naked
DNA &Liposome
1) Naked DNA
This is the simplest method of non-viral transfection,
intramuscular injection of a naked DNA plasmid have
occurred with some success; however, the expression has
been very low in comparison to other methods of
transfection. In addition to trials with plasmids, there have
been trials with naked PCR product, which have had
similar or greater success. This success, however, does
not compare to that of the other methods, leading to
develop more efficient methods for delivery of the naked
DNA such the use of a "gene gun", which shoots DNA
coated gold particles into the cell using high pressure gas.
2) Liposome
Liposome is an artificial lipid sphere with the therapeutic
DNA in the aqueous core it is capable of passing the DNA
through the cell membrane
Advantage of non-viral vector
• DNA/lipid complexes are easy to prepare
• there is no limit to the size of genes that can be delivered
• Carrier systems lack proteins; they may evoke much less
immunogenic responses.
• Much less risk of generating the infectious form or
inducing tumorigenic mutations because genes delivered
have low integration frequency and cannot replicate or
recombine.
Antisense therapy
Oligonucleotides
Is a form of treatment for genetic disorders or infections.
When the genetic sequence of a particular gene is known
to be cause of a particular disease, it is possible to
synthesize a strand of nucleic acid (DNA, RNA or a
chemical analogue) that will bind to the messenger RNA
(mRNA) produced by that gene and inactivate it,
effectively turning that gene "off
The use of synthetic oligonucleotides in gene therapy is to
inactivate the genes involved in the disease process. There
are several methods by which this is achieved. One
strategy uses antisense specific to the target gene to
disrupt the transcription of the defective gene.
Another strategy uses double stranded
oligodeoxynucleotides as a trick for the transcription
factors that are required to activate the transcription of the
target gene. The transcription factors bind to the it instead

Unit 2 - Molecular and genetic factors in disease
18
of the promoter of the defective gene, which reduces the
transcription of the target gene, lowering expression.
Problems with vectors:
1) The new gene might be inserted in the wrong location in
the DNA, possibly causing harmful mutations to the DNA
or even cancer.
2) 2.The possibility that transferred genes could
be overexpressed,producing so much of the
missing protein as to be harmful;
3) The viral vector could cause an immune reaction;
4) The viral vector could be transmitted from the patient to
other individuals or into the environment
• Current uses of gene therapy focus on treating or curing
existing conditions. In the future, the focus could shift to
prevention. As more of the human genome is understood,
medicine will know more about which genes contribute to
or cause disease. With that knowledge in hand, gene
therapy could be used to head off problems before they
occur
Is Gene therapy totally safe ???
Although gene therapy is a promising treatment option for
a number of diseases (including inherited disorders, some
types of cancer, and certain viral infections), the technique
remains risky and is still under study to make sure that it
will be safe and effective. Gene therapy is currently only
being tested for the treatment of diseases that have no
other cures
How can the patients receive: "the right medication, in
the right amount, in the right form, at the right time,
for the right disease
The answer is by the Personalized Molecular Medicine
Pharmacogenomics
Genetic testing for assessment of drug response may
predict the best specific drugs and dosages for individual
patients based on genetic profiling: so-called
‘personalized medicine’
Polymorphic mutations within genes can affect individual
responses to some drugs, such as loss-of-function mutations
Example:CYP2D6 gene is part of a large family of highly
polymorphic genes encoding cytochrome P450 proteins,
mostly expressed in the liver, which determine the
metabolism of a host of specific drugs. Polymorphisms in
the CYP2D6 gene determine codeine activation, while
those in the CYP2C9 gene affect warfarin inactivation
This. Polymorphisms in these and other drug genes
determine the persistence of drugs and, therefore,should
provide information about dosages and toxicity
Pathway medicine
The ability to manipulate pathways that have been altered
in genetic disease has therapeutic potential for Mendelian
disease, but a firm understanding of both disease
pathogenesis and drug action at a biochemical level is
required. An example of this has been the discovery that
the vascular pathology associated with Marfan’s
syndrome is due to the defect in fibrillin molecules
causing up-regulation of transforming growth factor
(TGF)- signalling in the vessel wall. Losartan is an
antihypertensive drug also acts as a partial antagonist of
TGF-signalling and is effective in preventing aortic
dilatation in Marfan’s syndrome

19

Unit 3 - Immunological factors in disease
20
Lecture 1 - Functional Anatomy &
Physiology of the immune system:
Functions of Host immune system:
1) To protect the host from pathogens while minimizing
damage to self-tissues.
2) Limits excessive responses that might lead to
autoimmune diseases.
Dysfunctions or deficiency of the immune response leads
to a variety of diseases involving every organ system in
the body.
The immune system consists of an intricately
Linked network of cells, proteins and lymphoid organs
which are strategically placed to ensure maximal
protection against infection.
Immune defenses are categorized into :
Innate & Adaptive immune response
Properties of immune responses
Innate immune responses
Adaptive immune resp.
Characteristics
1 recognize generic microbial
structure
Ag specific responses
2 immediate mobilized (minutes)
slow responses (days)
3 No memory
memory
4 Genetically encoded
Not genetically encoded
5 Identical responses in all
individuals
Aquired as an adaptive
immune response to
exposure to antigens
6 Present in invertebrates and
vertebrates
Presents in vertebrates
only
Immune components
1 Constitutive barriers (skin)
T and B lymphocytes
2 Phagocytes(N, Macro)
secreted molecules (Abs)
3 NK cell
Ag specific receptors
4 Soluble mediators
(Complement, Cytokines, acute
phase protein)
5 Pattern recognition molecules
(Toll like receptors)
The innate immune system
Constitutive barriers to infection
1) Skin:
Tightly packed highly keratinized cells of the skin which
limit colonization by microorganisms (MO).
Microbial growth is inhibited by physiological factors
such as low PH and low O2 tension and sebaceous glands
secrete hydrophobic oils that repel MO. Sweat contains
lysozyme that destroy bacterial cell wall. Ammonia has
antimicrobial activity. Defensine is also antimicrobial
peptides
2)
Mucous membrane
of the respiratory and
gastrointestinal and genitourinary tract provides a
constitutive barrier to infection.
Cilia and Secreted mucous trap invading pathogen.
Secretory IgA prevents bacteria and virus attaching to and
penetrating epithelial cells
Lysozyme and defensine
Lactoferrin acts to starve invading bacteria of iron
Physical manoeuvres such as sneezing and coughing
GIT: HCL, salivary amylase destroy bacteria
Induced vomiting or diarrhoea promotes clearance of
invading MO.
3) Endogenous commensal bacteria
About 10
14
bacteria normally reside at epithelial surface
in symbiosis with the human host and compete with
pathogenic bacteria for nutrients & space
They produce fatty acids and bactericidins that inhibits
growth of MO.
Eradication of these normal flora with broad spectrum
antibiotics commonly results in opportunistic infection
like candida
Phagocytes
They are specialized cells which ingest and kill MO and
produce inflammatory molecules which regulate other
components of the immune system
It includes Neutrophils, Macrophages & monocytes.
They are crucial for defense against bacterial & fungal
infections
They express a wide range of surface receptors to identify MO
These pattern recognition receptors include:
1- Toll-Like receptors
2- Mannose receptors
They recognize bacterial cell wall components, bacterial
DNA and Viral Double stranded RNA directly
Phagocytes recognize pathogen by these receptors alone.
Engulfment of microorganisms is greatly enhanced by
opsonisation.
Opsonins include acute phase proteins such as C reactive
protein, Abs, complement. This opsonin binds to pathogen
and phagocytes receptors acting as a bridge between the
two and facilitating phagocytosis

Unit 3 - Immunological factors in disease
21
Neutrophils
They are short lived cells with half-life of 6 hours
Kill MO directly
Facilitates the rapid transit of cells through tissues and
amplify the immune system
Killing mediated by enzymes contained in the granules
Changes in the damaged cells triggers inflammatory
molecules and cytokines to stimulate the production and
maturation of N in BM.
The transit of N. through the blood stream is responsible for
the rise in leukocyte count that occurs in early infection.
N. phagocytose the MO and fuse with cytoplasmic
granules to form phagolysosome.
Killing of MO ocuurs through a combination of oxidative
and non –oxidative pathways
1) Oxidative killing is also known as respiratory burst, is
mediated by NADPH oxidase enzyme complex which
converts oxygen into reactive oxygen species :
- Hydrogen peroxide - Superoxide
Myeloperoxidase enzyme
- Hypochlorous ions (HOCL
-
)
2) Non- oxidative (oxygen-independent killing)
Occurs through release of bactericidal enzymes including
lysozyme and lactoferrin
The process of phagocytosis deplete N glycogen reserves
and followed by N death
After N death, their content is released and lysosomal
enzymes degrade collagen & liquefaction of adjacent
tissues. The accumulation of dead and dying N. results in
the formation of pus. When the amount of pus is
extensive, result in abscess formation.
Monocytes and Macrophages
Monocytes are the precursors of tissue macrophages
They produced in the BM and exported to circulation
constitutes about 5% of leukocytes
After 7-10 Hrs in the blood stream , they migrate to
peripheral tissues where they differentiate into tissue
macrophages. They do not die after killing pathogens.
In the liver----- Kupffer cell
In the lung-----alveolar macrophages
In the kidney----mesangial cells
In the brain-----microglial cells
Functions of Macrophages:
1) Initiation and amplification of the inflammatory response
by cytokines secretions (IL-1, IL-6, IL-8, TNF alpha)
2) Killing of MO by phagocytosis (oxidative and non-
oxidative pathways)
3) Resolution and repair of inflammation by:
a. Scavenging of necrotic and apoptotic cell
b. Tissue remodelling by elastase and collagenase enzymes
c. Scar formation by IL-1 , platelet derived growth factor,
fibroblast growth factor
4) Link between innate and adaptive by presentation of Ag
to T cells and T cells secreted cytokines act on
macrophages that enhance phagocytosis.
Natural killer cells (NK) cell
Is large granular lymphocytes
Acts against tumor cell and virally infected cell
It is not Ag specific cell and generate memory cell
It kills target cell by Ab dependent cell mediated
cytotoxicity (ADCC) and perforin –granzyme pathway
Produce the following cytokines (TNF-α, IFN-α , IFN- γ)
Mast cell and Basophils
1) they are derived from BM 7 play an important role in allergy
2) Mast cell reside in the tissues that are exposed to external
stimuli like skin and gut
3) Basophiles reside in the circulation and recruited to
tissues in response to inflammation.
Both had a cytoplasmic granules containing histamine and
other mediators
Other mediators synthesized after activation
(Leukotrienes, prostaglandins)
Local release of these mediators initiates inflammatory
cascades that lead to increase local blood flow, vascular
permeability, stimulate smooth muscle contraction and
increase secretion at mucosal surface.
Cytokines
Are soluble proteins that acts as multipurpose chemical
messengers. More than 100 cytokines have been described
with overlapping complex roles in modifying the immune
response
IL-1 secreted from macrophages and stimulates N
recruitment, T cell and macrophages activation; induce
fever and acute phase reactant.
IL-2 secreted from Th 1 , stimulate proliferation and
differentiation of Ag specific T cell
IL-4 secreted from Th2 , and mast cell , stimulates
maturation of T and B cell and enhance IgE production
IL-6 secreted from Th2 and macrophages,stimulate
maturation of B cell to plasma cell
Il-12 secreted from macrophages and activate NK cell and
stimulates IFN gamma and TNF alpha release by T cell
Unit 3 - Immunological factors in disease
22
IFN alpha secreted from Th1 and macrophages and NK
cell and had antiviral activity & activates TCD8 cell, NK ,
macrophages
IFN gamma secreted from Th1cell that increases
antitumor and antimicrbial activity of macrophages
TNF alpha secreted from macrophages and it is pro
inflammatory and had direct cytotoxic, increase apoptosis
Complement system:
It is a group of more than twenty tightly regulated
functionally linked proteins that act to promote
inflammation and elimenate pathogens
It is synthesis in the liver
It is circulate in inactive form
It is activated by three pathways:
1) Classical pathway: is initiated when IgG or IgM
binds Ag forming immune complex
2) Alternative pathway: is initiated by binding C3 to
LPS of G –ve bacterial cell wall, C3 to teichoic acid of
G + ve bacteria
3) Lectin pathway: is activated by the direct binding of
mannose-binding lectin to microbial cell surface
carbohydrates(mannose on the pathogen surface)
This mimics the binding of C1 to immune complex, thus
bypassing the need for immune complex formation
Activation of the complement by any one of these
pathways results in activation of C3 ending in a final
common pathway
C5-C9 (MAC) membrane attack complex ending with
pores formation on the cell surface
It is important in defense mechanisms against encapsulated
bacteria (Neisseria & Haemophilus influenzae)
Fragments of complement act as an opsonin. naphylotoxins
The adaptive immune system:
When the innate immune system failed to eradicate the
pathogen the adaptive immunity will be stimulated
It is characterized by specificity, highly adaptive & can
respond to unlimited number of molecules & had a memory
There are two major arms of adaptive immunity:
1) Humoral immunity mediated by Abs which is produced
by B cells. 2) Cellular immunity mediated by T cells that
secrete cytokines
Lymphoid organs
Primary lymphoid organs that involved in lymphocytes
development which include BM where both T and B
lymphocytes are derived from haemopoietic stem cell and
where lymph.mature. The second organ is thymus
Thymus:
Is bilobed structure that active during fetal and neonatal
period and involuted after puberty. In the thymus
immature T cell undergoes selection and maturation.
Absence of thymus is associated with profound T cell
immune deficiency.
Secondary lymphoid organs include spleen, LN, mucosa
associated lymphoid tissue and gut associated lymphoid
tissue where the pathogen interacts with lymphocytes.
Spleen
:
Is highly effective in filtration of blood and important site
of phagocytosis of aged RBC, bacteria, immune
complexes and other debris
It is important defense against encapsulated bacteria
(Streptococcus pneumonia and H influenzae infection)
Lymph node
It consists from:
1) Cortex (B cell) primary follicles
2) Para cortex (T cell and dendritic cell)
3) Medulla (plasma cell) & contains sinuses hat rich in
macrophages
MALT
Includes Peyer’s patches, Appendix, tonsils & Adenoids
Lymphatics
Functions
1) Provide access to LN
2) 2-return tissue fluid to venous system
3) Transport fat from small intestine to the blood stream
It begins as blind ended capillaries & then form lymphatic
ducts then enter LN (Afferent) and leave LN (Efferent)
and drain into thoracic duct then superior vena cava
Humoral Immunity
B cells:
They arise in the BM stem cells and found as mature B
cells in BM, LN, spleen and to lesser extent blood stream.
It encountered soluble Ag usually in LN by Ab that found
on its surface and presented to T cells. Stimulated B cells
by Ag respond by proliferation known as clonal expansion
and differentiate into either plasma cell and memory cells
Immunoglobulins:
Are soluble proteins consists from two heavy and two
light chains. The heavy chain determins the isotypes IgG,
IgM, IgA, IGE,IgD
Functions: * Opsonins, ADCC
Unit 3 - Immunological factors in disease
23
* Actvation classical pathway of complement
* Present in the secretions (Sec IgA) and deficiency of
this leads to recurrent resp and GIT infection
First exposure to Ag leads to IgM production (longer lag
phase) and for short period of time
Second exposure to AG leads to large titer of IgG
production with higher affinity (shorter lag phase)
Cellular immunity:
T cells are important against virus ,fungi, intracellular
bacteria
It arise in the bone marrow and exported as immature
cells to thymus undergo differentiation and selection
Ag in order to recognized by T cells , must be processed
into smaller fragment and combined to HLA (human
leukocyte antigens)
HLA molecules are highly polymorphic to ensure
diversity in recognition of Ags within the population.
T cells can be divided according to its function,
recognition of HLA molecules and expression of cell
surface proteins.
Leukocytes cell surface molecules are named
systematically by assigning them a cluster of
differentiation (CD) Ag numbe
T cytotoxic (CD8)
They recognize Ag peptide in association with MHC class I
Kill target cell by perforin granzyme
Secret IFN gamma
T helper (CD4)
They recognize Ag peptide in association with MHC class II
Produce cytokines and provide co -stimulatory signals
that support the activation of Tc
Assist production of mature Ab by B cells
Th cells can be divided into subsets according to
cytokines they produced :
* Th1 (IL-2, IFN- gamma, TNF-alpha), support
development of delayed type hypersensitivity
* Th2 (IL-4, IL-5, IL-10) promote allergic response
* Regulatory CD4+ lymphocytes, important in the
regulation of other CD4 cells and prevention of
autoimmune diseases
Unit 3 - Immunological factors in disease
24
Lecture 2 - Immune deficiency
The consequences of deficiencies of the immune system
include the followings:
1- Recurrent infections.
2- Autoimmunity.
3- Susceptibility to malignancy.
Primary ID
Secondary ID
Due to infection, drug therapy, malignancy and ageing.
Phagocyte deficiency
Leads to increased bacteria and fungi infection.
Complement deficiency
Leads to infection: Neisseria meningitides, Neisseria
gonorrhoeae, Haemophilus influenzae & Streptococcus
pneumonia
T lymphocytes deficiency leads to:
Bacterial infection (TB), fungi infection (Candida), viral
infection (CMV) & Protozoa infection (pneumocystis carini)
Antibody deficiency leads to
1- Bacterial infection (Staphylococcus aureus)
2- Viral infection (Enterovirus)
3- Protozoa infection (Giardia lamblia)
Presenting problems in immune deficiency
Recurrent infections
Warning signs of ID:
1)
Eight respiratory tract infection/ year in a child or more
than four respiratory tract infection/ year in an adult.
2)
More than one infection requiring hospital admission or
intravenous antibiotics.
3) Infection with unusual organisms
4) Infection at unusual sites
5) Chronic infection unresponsive to usual treatment
6) Early end organ damage (Bronchiectasis)
7) Family history of immune deficiency
Investigations:
1-Full blood count 2- white cell differential
3- Acute phase protein (C-reactive protein)
4-Liver function test 5-Renal function test
6-Urine dipstick 7-Serum Immunoglobulin
8- Protein electrophoresis 9- Microbiological test.
10- Virological test. 11- Radiological test.
If ID is suspected, patients should not receive live
vaccines because of the risk of vaccine – induced disease.
Primary Phagocyte Deficiencies:
Usually present with recurrent bacterial and fungal
infections affecting unusual sites and majority present in
childhood but milder forms may present in adults.
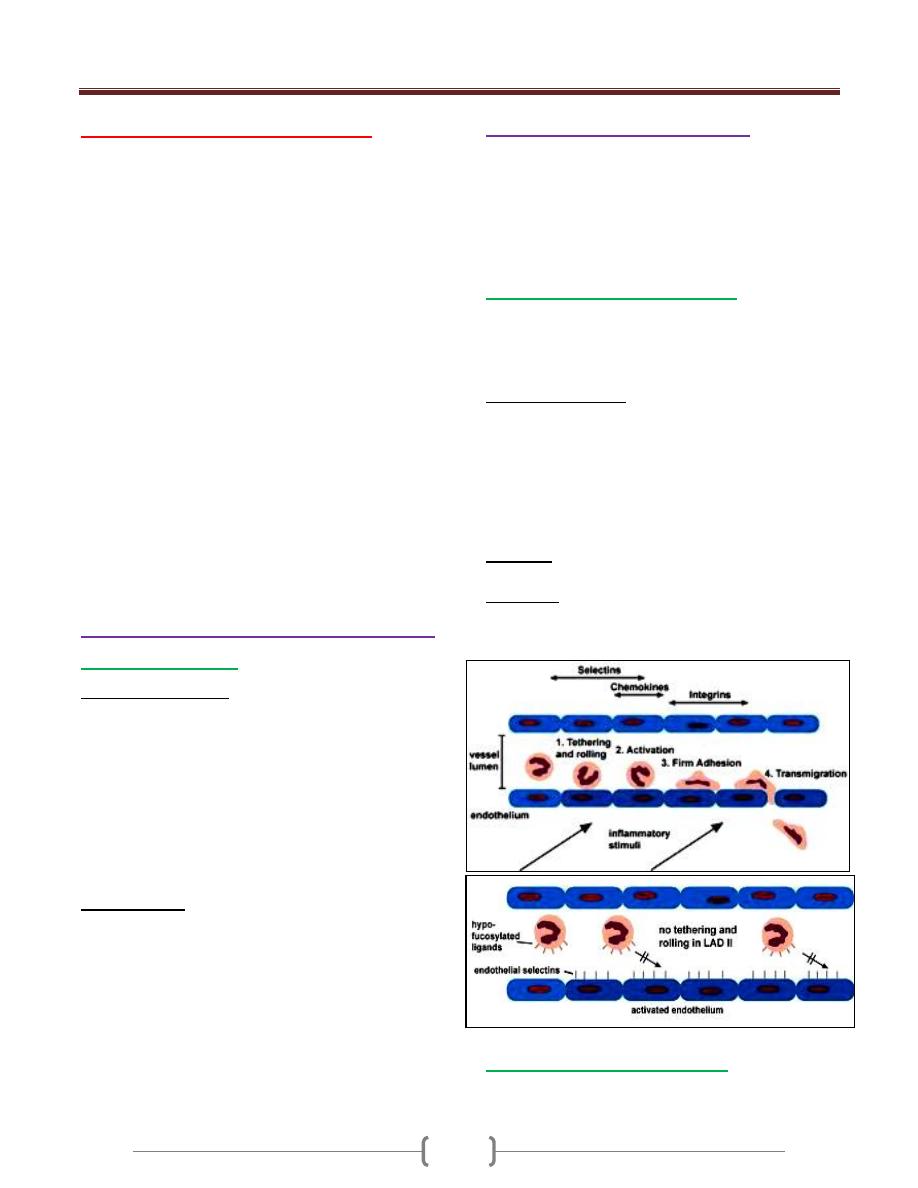
It includes: 1- Leukocyte adhesion deficiencies
2- Chronic granulomatous disease
3- Defects in cytkines and cytokines receptors
Leukocyte adhesion deficiencies:
It is an autosomal recessive disease.
These are disorders of phagocytes migration, where
failure to express adhesions molecules results in the
inability of phagocytes to exit the blood stream.
It is characterized by:
a) recurrent bacterial infections
b) Lack pus or neutrophils infiltration at site of infection
c) Peripheral neutrophils counts may be very high because of
the failure of mobilized N to exit blood vessels.
d) Infections are usually apparent from birth by presenting
infection is omphalitis with delayed separation of the
umbilical cord.
Diagnosis: by tests showed reduced or absent expression
of adhesions molecules on N.
Treatment: Prompt antibiotic therapy should be initiated
as early as possible in case of acute infection, bone
marrow transplantation and gene therapy.
Chronic granulomatous disease:
This results from mutations in the genes encoding the
NADPH oxidase enzyme, causing a failure of oxidative

Unit 3 - Immunological factors in disease
25
killing. This may be demonstrated using the nitroblue
tetrazolium reduction test
The defect leads to susceptibility to catalase – positive
organisms such as Staphylococcus aureus
Intracellular killing of mycobacteria in macrophages is
also impaired.
Infections most commonly involve the lungs, lymph
nodes, soft tissues, bone, skin and urinary tract.
People with this condition often have areas of inflammation
(granulomas) in various tissues that can be damaging to
those tissues
Defects in cytokines and cytokines receptors:
Defect in cytokines such as IFN-γ, IL-12 or their receptors
results in failure of intracellular killing
Individuals are susceptible to mycobacterial infections
Treatment:
1- Intravenous antibiotics for treatment existing infection.
2 -surgical drainage of abscess.
3- Long term prophylaxis with antifungal agents
4- Specific treatment depends upon the nature of defect
and stem cell transplantation may be considered.
Complement pathways deficiencies
1) Genetic deficiency of classical complement pathway (C1,
C2, C4) are associated with high prevalence of
autoimmune disease particularly systemic lupus
erythematosus (SLE)
2) Deficiency in C9 increase Neisseria species infection
(Gonococcal & Meningococcal) and encapsulated bacteria
3) Deficiency of Mannose - binding lectin leads to increased
incidence of bacterial infection if subjected to additional
cause of immune compromise such as prematurity or
chemotherapy.
However, the important of this deficiency is not important
in healthy individuals.
Investigations and treatment:
1- Complement C3 and C4 measurements
2- CH50 test (Classical haemolytic pathway 50) or called
THC (Total Hemolytic Complement)
Sheep RBC coated with Abs + patient’s serum leads to
complete lysis RBC
There is no definitive treatment.
Patients should be vaccinated with meningococcal,
pneumococcal and H. influenzae vaccines in order to
boost their adaptive immune response.
Lifelong protective Penicillin to prevent infection
Family members should be screened for complement
deficiency.
Primary deficiencies of the adaptive
immune system
Combined B and T lymphocytes deficiency:
It is due to defect in lymphoid precursors
Results in combined failure of B and T cell Maturations
Cause recurrent bacterial, fungal and viral infections soon
after birth
Treated by stem cell transplantation or gene therapy still
under investigations.
Primary T lymphocytes deficiency
Characterized by recurrent viral, protozoal and fungal
infections
It is associated with defective Abs productions because of
the importance of Tcell in providing help to B cells
These disorders present in childhood
It includes the following diseases:
a- DiGeorge Syndrome b- Bare Lymphocytes Syndrome
c- Auto Immune Lymphoproliferative Syndrome
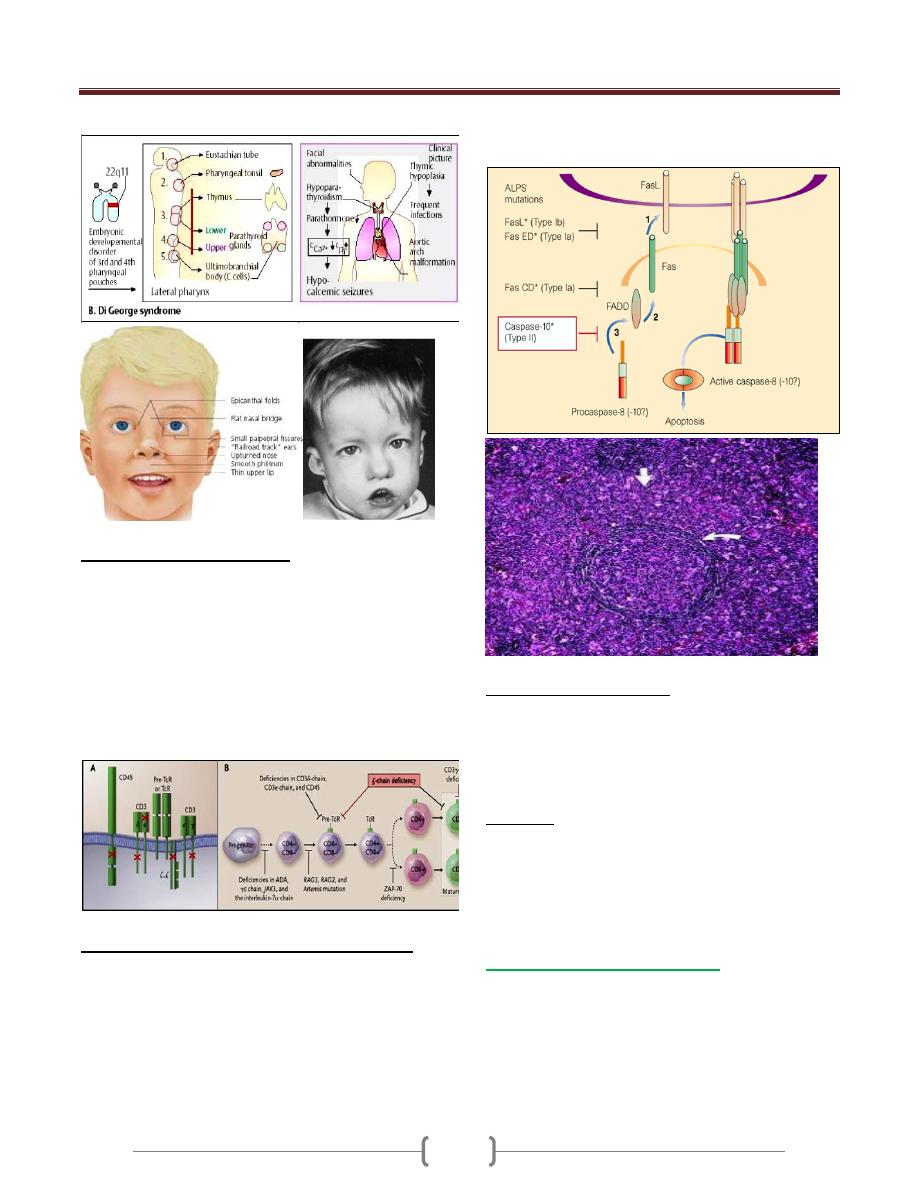
DiGeorge Syndrome
Results from failure of development of the 3
rd
and 4
th
pharyngeal pouch, usually caused by deletion of a small
piece of chromosome 22 at a location designated 22q11.2
produced by an error in recombination at meiosis
It is a congenital thymic aplasia & thymic hypoplasia,
It is associated with abnormalities of the aortic arch,
hypocalcaemia, tracheo-oesophageal fistula,
Cleft lip and palate and absent thymic development.
Characterized by very low numbers of T cells despite the
normal development in the bone marrow
Unit 3 - Immunological factors in disease
26
Bare Lymphocytes Syndrome
Characterized by absent expression of HLA molecules
within the thymus.
If HLA class I affected, CD8 + lymphocytes fail to
develop
If HLA class II affected, CD4 + lymphocytes fail to
develop
Uncontrolled activation of NK cell due to absence of
HLA class I expression
leads to recurrent infection and systemic vasculitis
Auto Immune Lymphoproliferative Syndrome:
Characterized by accumulation of lymphocytes and
persistence of autoreactive cells. Unusually high numbers
of white blood cells called lymphocytes accumulate in the
lymph nodes, liver, and spleen, which can lead to
enlargement of these organs
Caused by failure of apoptosis. This condition is usually
caused by mutations in the FAS gene
Patients develop lymphadenopathy, splenomegaly and a
variety of autoimmune diseases.
Investigations and treatment:
1) Measurement of total T- lymphocytes count
2) Measurement of T lymphocytes subsets
3) Immunoglobulin measurements
4) functional tests of T cells activation and proliferation
5) HIV test may be indicated
Treatment
1) Patients should receive anti-Pneumocystis and antifungal
prophylaxis treatment
2) Immunoglobulin replacement if disease associated with
defective Ab production.
3) Stem cell transplantation 4) Thymic transplantation
Primary Antibodies deficiency:
Characterized by recurrent bacterial infections.
Particularly of the respiratory and gastrointestinal tract.
The most causative organisms are bacteria such as
Streptococcus pneumonia and H influenzae .
Usually present at 5-6 months of age when the protective
benefit of transferred maternal Igs has decreased.

Unit 3 - Immunological factors in disease
27
3 major primary Abs deficiencies present in adulthood
1) Selective IgA deficiency
Characterized by low or undetectable IgA.
Most common primary Immune deficiency.
In some patients, there is a compensatory increase in
serum IgG levels.
30% of individuals experience recurrent mild respiratory
and GIT infections.
2) Common variable immune deficiency
It is a heterogeneous adult-onset primary immune
deficiency of unknown cause.
Characterized by low serum IgG levels and failure to
make Abs responses to exogenous pathogens.
Paradoxically, Ab mediated autoimmune diseases like
autoimmune hemolytic anemia
It is also associated with increased frequency of
malignancy like lymphoprolifrative diseases.
3) Specific Ab deficiency or Functional IgG Ab deficiency
It causes poor Ab responses to polysaccharide Ags.
Some patients are deficient in the Ab subclasses IgG2 & IgG4
It is previously called IgG subclass deficiency
Investigations:
1) Measurements of serum Igs.
2) Protein electrophoresis
3) Urine electrophoresis to exclude secondary causes of
hypogammaglobulineamia
4) Specific Ab responses to specific pathogens , if it is low ,
vaccinate the patient with killed vaccine
5) Quantitation of T and B lymphocytes
Treatment:
1) Treatment of infections by antibiotics and prophylaxis
antibiotics may be indicated.
2) Ig replacement intravenously which derived from pooled
plasma & contains IgG Abs & administered every 3-4 weeks
** Vaccinations with live vaccines is contraindicated
Secondary Immune deficiencies
It is more common than primary immune deficiency
Causes
1) Physiological: aging, prematurity, pregnancy.
2) Infection: HIV, measles, TB
3) Iatrogenic: Drugs like immunosuppressive drugs,
corticosteroids, antineoplastic, radiotherapy
4) Malignancy: Leukemia, lymphoma, myeloma, solid tumor
5) Biochemical and nutrional disorder:
6) Others: Burns, Asplenia.
Immune senescence:
It is a decline of the immune response in the elderly
characterized by:
1) Decline in the T cell response with reduced delayed type
hypersensitivity reaction.
2) Decrease in Abs production for many exogenous pathogens
3) AutoAbs rise but autoimmune diseases are less common
4) Reduced responses to vaccinations, about 30% of healthy
older people may not develop protective immunity after
influenza vaccine.
5) Allergic disorders and transplant rejection is less common
6) Increased susceptibility to infections like respiratory tract
infection, UTI, latent infections like TB and Herpes
Zoster may be reactivated.
7) Absent manifestations of infections e.g. leukocytosis &
pyrexia.
Unit 3 - Immunological factors in disease
28
Lecture 3 - The Inflammatory
Response
Inflammation is the response of tissues to injury or
infection and is necessary for normal repair and healing
Physiology & pathology of inflammation
Acute inflammation
It is the result of rapid and complex interplay between the
cells and soluble molecules of the innate immune system.
When there is an infection or inflammation in the tissue,
leads to infiltration of phagocytic cells (N, Macro) & release:
1) Enzymes (Cyclo-oxygenase & nitric oxide synthase). It
leads to release (prostaglandins, histamine, kinins,
anaphylotoxins & nitric oxide)
2) Cytotoxines (IL1, TNF, IL6)
This leads to:
Vasodilation, increased vascular permeability, increased
production of N in the bone marrow, release of insulin
from pancreas, release glucocorticoids & catecholamines
from adrenal, increase heart rate, increase synthesis of
acute phasr protein and amyloid A by the liver, low blood
pressure, fever, enlarged lymph nodes and fibrinogen
plays important role in wound healing.
Control mechanisms of inflammation
Acute phase proteins
α1-antitrypsin and α1-antichymotrypsin control the pro-
inflammatory cascades.
Antioxidants such as haptoglobin and manganese
superoxide dismutase scavenge for oxygen free radicals.
Increased iron-binding proteins such as transferrin, ferritin &
lactoferrin decrease the iron available for uptake by bacteria.
Resolution of inflammation
This involves active down-modulation of inflammatory
stimuli and repair of bystander damage to local tissues.
1) Extravasated neutrophils undergo apoptosis and are
phagocytosed by macrophages, along with the remains of
microorganisms.
2) Macrophages also synthesise collagenase and elastase,
3) Macrophage-derived cytokines, including transforming
growth factor (TGF)-β and platelet-derived growth factor,
attract fibroblasts and promote the synthesis of new collagen
4) Angiogenic factors stimulate new vessel formation.
Sepsis and septic shock
Septic shock is the clinical manifestation of overwhelming
inflammation. Failure of normal inhibitory mechanisms
results in excessive production of pro-inflammatory
cytokines by macrophages, causing hypotension,
hypovolaemia, decreased perfusion & tissue oedema.
Damage to the vascular endothelium and further
increasing capillary permeability.
Direct activation of the coagulation pathway ends with
clot formation.
The clinical manifestations:
1) Cardiovascular collapse
2) Acute respiratory distress syndrome
3) Disseminated intravascular coagulation
4) Multi-organ failure and often
5) Death
Cause: infection with Gram-negative bacteria, because
lipopolysaccharide is particularly effective at activating
the inflammatory cascade.
Chronic inflammation
Failure to remove an inflammatory stimulus.
Persisting microorganisms stimulate the ongoing
accumulation of neutrophils, macrophages and activated T
lymphocytes & deposition of fibrous connective tissue
ending with granuloma.
Eg, (TB, Leprosy) because this MO is protected with
thick cell wall,
Investigations in inflammation
1) Complete blood picture: Leukocytosis
2) Platelets count: may be increased
3) Erythrocyte sedimentation rate (ESR)
4) Blood film: normocytic normochromic anemia
5) C-reactive protein & complement: increased
6) Albumin: reduced
7) Plasma viscosity
C-reactive protein
Is an acute phase protein synthesis in the liver.
It is opsonizes invading pathogens.
It’s level increased within 6 hours of an inflammatory
stimulus.
It indicates acute inflammation
Its half-life = 19 hours
It is used to monitor disease activity.
Erythrocyte sedimentation rate (ESR)
It is an indirect measure of acute phase protein.
It measures the rate of fall of RBC through plasma &
aggregation of erythrocytes
Unit 3 - Immunological factors in disease
29
Normally RBC do not aggregate with each other because
of their repellent negative charge.
Plasma protein is positive charge & act to neutralize the
surface charge of RBC.
When there is an increase in plasma protein particularly
fibrinogen, overcome the repulsive forces causing stack of
RBC (rouleaux)
It is measuring plasma protein composition, concentration
& RBC morphology.
It is increased (1) in acute inflammation, when there is an
increase in acute phase protein. (2) when there is an
increase in IgG, IgM & IgA.
It is deceased when there is an abnormality in RBC
morphology like spherocytosis, sickle cell anemia.
Plasma viscosity
It measures plasma protein concentration
It is affected by concentration of large plasma proteins
(fibrinogen, Ig [IgM])
Presenting problems in inflammation
Unexplained raised ESR
The ESR should not be used as a screening test for the
presence of disease in asymptomatic patient.
Clinical assessment
There is an extreme elevation of ESR ˃ 100 mm/hr in the
absence of significant disease.
Investigations
CRP, Serum Ig, Urine electrophoresis
Full blood count; anemia, leukocytosis, neutrophilia,
abnormal lymphocytes
Liver function test
Blood & Urine cultures
Imaging: Chest X-ray, Abdominal CT scan, Abdominal &
pelvic ultrasound, MRI, Echocardiography and Isotope scan
Periodic fever syndromes
These rare disorders are characterized by recurrent
episodes of fever and organ inflammation associated with
an elevated acute phase response.
Familial Mediterranean fever
This is the most common of the familial periodic fevers,
predominantly affecting Mediterranean people, including
Arabs, Turks, Sephardic Jews and Armenians.
It results from mutations in pyrin gene.
Characterized by painful attacks of fever associated with
peritonitis, pleuritis and arthritis, and lasts from a few
hours to 4 days.
CRP is elevated.
Colchicine is the treatment of choice.
Hyper IgD syndrome
It is characterized attacks of fever, abdominal pain,
diarrhoea, lymphadenopathy, arthralgia, skin lesions and
aphthous ulceration.
Laboratory features include an acute phase response
(elevated CRP & ESR) and markedly elevated IgD
It has mainly been described in the Netherlands & France.
It is AR disease.
All patients with syndrome have mutations in the gene for
mevalonate kinase which is involved in cholesterol
metabolism.
No specific treatment is available.
TNF receptor associated periodic syndrome
Also known as TRAPS or familial Hibernian fever.
Is a periodic fever syndrome associated with mutations in
a receptor for the molecule tumor necrosis factor (TNF)
that is inheritable in an autosomal dominant manner.
Individuals have episodic syndromes such as recurrent
high fevers, rash, abdominal pain, joint/muscle aches &
puffy eyes.
It causes recurrent episodes of fever that typically last 1-3
weeks that are associated with chills & severe muscle pain
in the trunk & the arms.
Patients develop a red & painful rash from the trunk to the
arms & legs, abdominal pain with nausea, vomiting &
diarrhea are common, as are red, swollen eyes. Other
important features include chest pain due to inflammation
of the membrane surrounding the lungs or heart.
Laboratory findings
1) Neutrophilia
2) Increased CRP
3) Elevated ESR
4) Low level of serum soluble type 1 TNF receptor
5) Molecule analysis of TNFRSF1A gene
6) Screening for proteinuria
Several medications have been studied for the treatment
of TRAPS including: Corticosteroids, Infliximab, Soluble
TNF receptor therapy.

Unit 3 - Immunological factors in disease
30
Amyloidosis
It is a disorder resulting from abnormal & insoluble
protein (amyloid) deposits extracellular in body tissues.
It consists of fibrils of specific protein linked to glucose-
aminoglycans, proteoglycans & serum amyloid protein.
It is either hereditary or acquired.
It is either localized or systemic.
Hereditary systemic amyloidosis
It is systemic amyloidosis.
Production of abnormal protein with abnormal structure.
It is AD due to mutation in transthyretin retinol
Transthyretin (TTR) is a serum & cerebrospinal fluid
carrier of the thyroid hormone thyroxin (T4) and retinol –
binding protein bound to retinol. This is how transthyretin
gained its name, transports thyroxine & retinol. The liver
secretes transthyretin into the blood & the coroid plexus
secretes TTR into the cerebrospinal fluid.
Characterized by neuropathy, cardiomegaly & renal
involvement
Acquires systemic amyloidosis
1) Reactive amyloidosis
This is the result of another chronic inflammatory disease
such as lupus, rheumatoid arthritis, tuberculosis,
inflammatory bowel disease (Crohn’s disease & ulcerative
colitis) & certain cancers.
It most commonly affects the spleen, kidneys, liver,
adrenal gland & lymph nodes.
AA means the amyloid type A protein causes this type of
amyloidosis.
90% of the patients developed non-selective proteinuria or
nephrotic syndrome.
2) Light chain amyloidosis
AL occurs when a specialized cell in the bone marrow
(plasma cell) spontaneously overproduces a particular
protein portion of an antibody called the light chain. (This
is why it is coded as AL)
Can occur with a bone marrow cancer of plasma cells
called multiple myeloma, Monoclonal gammmaopathy.
Requiring chemotherapy treatment.
It is characterized by neuropathy, cardiomyopathy,
proteinuria, amyloid nodules.
3) Dialysis associated amyloidosis
Hemodialysis-associated amyloidosis is a form of
amyloidosis associated with chronic renal failure.
Long-term hemodialysis results in a gradual accumulation
of β
2
microglobulin, a serum protein in the blood. It
accumulates because it is unable to cross the dialysis filter
Β
2-
micrglobulin is a major constituent of amyloid fibrils.
It has been shown that through accumulation, it invades
synovial membranes & osteoarticular sites. As a result, it
causes destructive osteoarthropathies such as
4) Senile systemic amyloidosis
Age ˃ 70 years
Normal transthyretin protein deposited in tissues.
Usually asymptomatic.
Affect ˃ 90% of 90 years old persons.
Diagnosis
A tissue sample of abdominal wall fat, the rectum or a
salivary gland can be examined in biopsy for evidence of
characteristic amyloid deposits.
The tissue is treated with various stains. The most useful
stain in the diagnosis of amyloid is Congo red, which,
combined with polarized light makes the amyloid proteins
appear apple-green on microscopy.
The nature of the amyloid protein can be determined by
various ways: the detection of abnormal proteins in the
bloodstream (on protein electrophoresis or light chain
determination), binding of particular antibodies to the
amyloid found in the tissue.
Treatment
Chemotherapy is the first line treatment in AL with
nephalan plus dexamethasone.
In AA, symptoms may improve if the underlying
condition is treated.
In familial causes of amyloidosis, a liver transplant can be
curative.

Unit 3 - Immunological factors in disease
31
Lecture 4 - Autoimmune disease
Is the presence of immune responses against self-targets.
It is identified by the presence of low titer autoantibodies
or autoreactive T cells.
Caused by failure of immunological tolerance:
Physiology & pathology of autoimmunity
Immunological tolerance
Tolerance
Autoimmune diseases
1 clonal deletion in
bone marrow and
thymus
Some autoreactive cells inevitably
evade deletion and escape to
peripheral circulation
2 suppression of
autoreactive cells by
regulatory T cells
generation of hyporesponsevness
(anergy) in lymphocytes which
encounter the antigens in the
absence of costimulatory signals
3 privileged sites (eye)
released Ags from these sites
Factors predisposing to autoimmune diseases
1) Genetic factor:
HLA genes (B27 association with ankylosing spondylitis,
type I diabetes associated with DR3/DR4, Myasthenia
gravis DR3
Genes determining cytokines activity, costimulation &
cell death
2) Environmental factor:
a) Infection with microbs
Eg. Acute rheumatic fever following streptococcal
infection, Reactive arthritis following bacterial infection
This is due to cross reactivity ( molecular mimicry),
Release of sequestered Ags, production of
inflammatory cytokines that leads to tissue damage
b) Drugs (Halothane, methyl dopa)
3) Sex:
Female more affected than males.
Classification of autoimmune diseases
•
It is classified as:
1) Organ specific diseases
2) Non –organ specific (Multisystem) diseases
There is some overlapping between them
The predominant mechanisms in tissue damage is type II,
III and IV hypersensitivity reactions
Mechanisms of tissue damage n autoimmunity:
1) Type II HS: binding of cytotoxic IgG and IgM to cell
surface causes cell killing
2) Type III HS: IgG or IgM bind soluble Ags to form immune
complexes which trigger classical complement pathway
3) Type IV HS: activated T cells, NK, phagocytes
Investigations in autoimmunity
a) Detection of specific autoantibodies in the patient’s
serum. The Ab is quantified either by titer ( minimal
dilution at which the Ab can be detected or by
concentration in standardized unit)
- This includes:
1- Rheumatoid factor 2- anti CCP Ab
3- Anti nuclear antibody 4- Abs to extractable nuclear Ags
5-Anti DNA antibodies 6-Antiphospholipid antibodies
7- Anti neutrophils cytoplasmic antibodies
b) measurement the complement components
Autoantibodies
RF
It’s an autoAb director against FC region of human IgG
It may be any Ig class but IgM is most commonly tested
In general a titer > 1:40 is considered positive
50% of patients with RA are + for RF at the time of
diagnosis. 25% will become seropositive in the first two
years of disease Thus it is insensitive to rule out RA at the
time of diagnosis
It has low specificity for RA because it is associated with
other conditions:
1) SLE
2) TB
3) elderly >65 ys
4) RA with extra –articular manifestation
5) Sjogen’s syndrome
6) mixed essential cryoglobulinaemia
7) primary billiary cirrhosis
The major indication for RF testing is to evaluate
prognosis in RA
When it is positive, it is associated with more sever
erosive disease and extra –articular disease manifestations
such as nodule, vasculitis.
Anti-CCP antibody
Abs to CCP (cyclic citrullinated peptide)
In this peptide, aa converted to
It is more specific test than RF for RA
It is a better predictor of an aggressive disease course
Antinuclear antibodies (ANA)
Are group of Abs which bind to components of the nucleus
Titer >1:80 is usually considered positive

Unit 3 - Immunological factors in disease
32
It is positive in (SLE, Scleroderma, dermatomyositis,
mixed connective tissue disease, autoimmune hepatitis,
5% of healthy individuals have an ANA titer >1:80
It is not useful in the diagnosis (RA, autoimmune thyroid
disease, malignancy and infectious disease)
Repeating ANA is not useful, no role for serial monitoring
of ANA titer
No correlation with disease activity
Abs to extractable nuclear Ags(ENA)
When ANA is positive , it is useful to establish which
nuclear component is being recognize
Some nuclear Ags are soluble and can be extracted from
the nucleus
There is little value in testing for ENA if the ANA is negative
It’s include Abs to (histone, centromere, smith, RNA
polymerase I)
Anti DNA Abs
Abs to single strand DNA is not specific
Abs to Double strand DNA is highly specific for SLE (95%)
Very high titers are associated with more sever disease
including renal and CNS involvement in SLE
They are useful in disease monitoring as an increase in Ab
titers is associated with disease activity and may precede
disease relapse
Antiphospholipid Abs (APL)
Are associated with the development of venous and
arterial thrombosis and recurrent fetal lose
It may be either: primary or secondary
It may be associated with SLE , malignant conditions,
infections and rheumatic conditions
There are sevaral kinds of APL Abs (anticardiolipin and
lupus anticoagulant)
Anticardiolipin Abs are Igs directed against phospholipid
particularly β2 glycoprotein-1.
Lupus antigoagulant Abs are Igs directed against
prothrombin and occasionally β2 glycoprotein-1.
They have overlapping specificity, if there is a clinical
suspicions of APLsyndrome, both tests should be performe
Lupus antigoagulant Abs should not be done when the
patients is on anticoagulant therapy.
Anti-neutrophil cytoplasmic antibodies (ANCA)
It is an IgG Abs directed against cytoplasmic constituents
of granulocytes.
It is of two types:
1) Cytoplasmic (c-ANCA): Abs to proteinase -3 associated
with Wegener’s granulomatosis
2) Pernuclear (p-ANCA): Abs to myeloperoxidase,
lactoferrin & elastase, it is associated with microscopic
polyarteritis
Atypical p-ANCA which are not due to myeloperoxidase
are commonly found in patients with ulcerative colitis and
autoimmune liver disease.
Serial measurement of anti-PR3 or anti –MPO antibodies
may be useful for disease monitoring.
Measurement of complement activity
Quantitation of complement components (C3 and C4),
may be useful in the evaluation of immune complex
mediated diseases (SLE)
Cryoglobulinaemia
Cryoglobulins are Ig that form precipitates in the cold
It is classified into 3 types: Type I, Type II & Type III
Testing for this Cryoglobulins requires the transport of a
serum to the laboratory at 37C0
Type I
Monoclonal IgM paraprotein
It is associated with lymphoproliferative disease
Symptoms: Rayanaud’s phenomenon , retinal vessel
occlusion, arterial and venous thrombosis
Protein electrophoresis done for detection IgM
monoclonal Ab
Serum viscosity raised
Type II
Monoclonal IgM paraprotein directed against IgG
Associated with infections (HBV, HCV)
Symptoms: small blood vessels vasculitis, purpuric rash ,
arthralgia ,hepatosplenomegaly, coetaneous ulceration, :
Rayanaud’s phenomenon
RF strongly positive, Decreased C4
Protein electrophoresis done for detection IgM
monoclonal Ab
Type III
Polyclonal IgM or IgG directed towards IgG
Associated with infection (HBV,HCV) SLE, RA
Symptoms: small blood vessels vasculitis, purpuric rash ,
arthralgia ,hepatosplenomegaly, coetaneous ulceration, :
Rayanaud’s phenomenon
RF strongly positive, Decreased C4
No monoclonal paraprotein
Management includes avoidance of cold and treatment of
underlying pathology
Type II and III need immunosuppression and or
plasmapheresis to remove the pathogenic Abs

Unit 3 - Immunological factors in disease
33
Lecture 5 -
Allergy
Allergic diseases are a common & increasing cause of illness
Affecting 15-20% of the populations
Atopy is the tendency to produce an exaggerated IgE
immune response to harmless environmental substances
Pathology of allergy
Normally the immune system does not make detectable
response to many environmental substances like food and
inhaled particles to which it is exposed on a daily basis
In allergy: exposure to allergen leads to production of
specific IgE Abs that bound to surface mast cells
Upon re-exposure, the allergen binds to membrane bound
IgE which activates the mast cell
•
This leads to release mediators causing either:
1-early phase response (sneezing and rhinorrhea)
2- Late phase response which occur after 4-8 hours
characterized by persistence swelling and inflammation
- Long standing or recurrent allergic inflammation may
give rise to a chronic inflammatory response
Factors influencing susceptibility to allergic diseases
Hygiene hypothesis: infections in early life bias the
immune system against the development of allergy
Positive family history
Genetic factor (genes that control cytokins production and
IgE level)
Presenting problems in allergy
A general approach to the allergic patient
Common presentation of allergic diseases
- Urticaria - Angioedema
- Atopic dermatitis - Allergic conjunctivitis
- Allergic rhinitis (hay fever) - Asthma
- Food allergy (egg) - Drug allergy
- Allergy to insect venom - Anaphylaxis
Clinical assessment
It is important to identify what the patient means by allergy.
Up to 20% of the UK population describe themselves as
having a food allergy
< 1% have an IgE mediated HS reaction
Enquire about allergic symptoms (past, present)
Family history of allergic diseases
Identify potential allergens in the home and workplace
Investigations
1) Skin prick test : read after 10 mins and a positive result
is indicated by a local wheal and flare response ≥ 2 mm
larger than negative control
2) Specific IgE test (RAST)radio allergo sorbent test:
quantitation IgE specific to allegen
3) Supervised exposure to allergen (challenge test):
bronchial provocation test and done in special center
4) Mast cell tryptase : measurement of this enzyme in the
serum , peaks at 1-2 hrs remaining elevated for 24 hrs
Nonspecific markers of atopic diseases
A. Total serum IgE: is elevated in atopic diseases , parasites
and helminthes infection, lymphoma
- Significant allergic diseases can occur despite a normal
total IgE level
B. Peripheral blood eosinophilia : is common in atopic
individuals and >20%
Treatment
Avoidance of allergen
Antihistamines block histamine H1 receptors. S.e.
sedation
corticosteroids: down- regulates proinflammatory
cytokines production
Sodium cromoglicate : stabilizes mast cell membrane and
inhibit the release of vasoactive mediators. It is used
prophylaxis
Ag –specific immunotherapy: by sequential
administration of escalating amount of diluted allergen
over a prolonged period of time
Omalizumab: monoclonal Ab against IgE
Preloaded self-injectable adrenaline : used in anaphylaxis
Anaphylaxis
It is a life threatening, systemic allergic reaction caused
by release of histamine and other mediators.
It is either
IgE mediated mast cell degranulation (Causes)
1) Food (Fish, egg, peanut)
2) 2-insect stings (Bee, Wasp)
3) 3-chemicals (penicillin, latex)
Non –IgE mediated mast cell degranulation
(anaphylactoid) (Causes)
1) Drugs (Aspirin, radiocontrast media)
2) Physical (Exercise, cold)
3) Idiopathic
Clinical assessment (manifestation)
1) Flushing and sweating
2) Wheeze due to bronchoconstrictions

Unit 3 - Immunological factors in disease
34
3) Hypotension
4) Urticaria
5) Cardiac arrhythmia
6) Angioedema of lips and mucous membrane
7) Laryngeal obstruction (strider)
8) Diarrhoea and abdominal pain
9) Loss of consciousness
Differential diagnosis
1) Causes of loss of consciousness ( myocardial infarction,
vasovagal syncope)
2) Causes of respiratory distress (status asthmaticus )
3) Causes of laryngeal obstructions (C1 inhibitor deficiency)
4) Causes of generalized flushing ( Carcinoid Syndrome)
5) Other causes (phaeochromocytoma)
Investigations
Serum mast cell tryptase
Specific IgE is important in anaphylaxis
Treatment
Prevention further exposure to allergen
Ensuring patent airway patency
Administration of oxygen
Restoration of blood pressure (laying the patient flat and
iv fluids)
Prompt administration of adrenaline that reverse the
action of histamine, given im ( adult dose 0.3 ml , 300
micrograms 1:1000 solution and repeated at 5-10 minutes
intervals if the initial response is inadequate
Intravenous antihistamine (chlorphenamine 10-20 mg im
or slow iv injection)
Corticosteroids (100-300 mg) prevent late –phase
symptoms in severly affected patients
Nebulised β2 –agonists may also be indicated
Angioedema
•
Is the episodic, localized, non –pitting swelling of
submucous or subcutaneous tissues.
•
It may occur alone or in conjunction with urticaria
•
It is a result of mast cell degranulation which may be
spontaneous or triggered by an allergic IgE mediated
response.
•
It is caused by drugs (aspirin, ACE inhibitors, NSAID,
radiocontrast media, antibiotics)
Clinical presentations
Localized soft tissue oedema (face, extremities, genetalia)
Respiratory tract obstruction (larynx, tongue)
Oedema of the intestine (abdominal pain , distention)
It is usually accompanied by urtcaria
It is discriminate from hereditary angioedema is not
associated with urticaria, does not respond to
antihistamine therapy.
Investigations
- Full blood count - CRP
- Thyroid function test - Liver function test
- Skin prick test - Specific IgE test
- C3, C4, C1 inhibitor level
Treatment
Oral antihistamines
Urgent medical attention when there is tongue or throat
swelling and fatal airway obstructions.
Specific allergies
Insect venom allergy
Local reactions to insect stings may cause extensive
swellings around the site lasting 7 days
Peanut allergy
Is most common food related allergy. It is rarely resolve
and lifelong avoidance is recommended
C1 inhibitor deficiency
Hereditary angioedema (HAE)
(inherited C1 inhibitor deficiency)
It is AD disorder due to decreased production of C1
inhibitor protein
C1 inhibitor is complement regulatory protein that inhibit
classical pathway of the complement
It is also a regulatory protein for the kinin cascade
It may be spontaneous or triggered by local trauma or
infection
Edema of the face, extremities, upper airway (laryngeal
obstructions) and GIT
Episodes of HAE are self- limitings and usually resolve
within 48 hours.
Acute episodes are accompanied by low C4 levels
Diagnosis by low level of C1 inhibitor
Treatment with infusion of purified C1 inhibitor
preparations and fresh frozen plasma
Acquired C1 inhibitor deficiency
Clinically indistinguishable from HAE
Associated with autoimmune & lymphoprolifrative disease
Treatment of underlying disorder may induce remission of
angioedem

Unit 3 - Immunological factors in disease
35
Transplantation and graft rejection
•
It is a definitive treatment of end stage organ disease
•
Major complications is graft rejection and infection
The complications depend on:
1) Type of organ transplanted
2) Genetic disparity
3) Primary disease
4) Drug therapy used
Transplant rejection
Solid organ transplantation stimulates an aggressive
immune response by the recipient, unless the transplant is
between monozygotic twins
The most important genetic determinant is the difference
between donor and recipient HLA proteins
Compatibility at all HLA loci decrease acute rejection
improves graft survival and allows the use of less intense
immunosuppressive protocols.
Types of rejection
1) Hyperacute rejection
Results in rapid and irreversible destruction of the graft
It is mediated by pre-existing recipient Abs against donor
( ABO, HLA) which is arise due to previous exposure to
transplantation, blood transfusion and pregnancy
It is type II HS reaction, end with thrombosis and necrosis
No treatment
2)
Acute rejection
Occur after 5-30 days
Treatment by increasing immunosuppressive drugs
a. Acute vascular rejection: it is due to Abs formation and
T, B lymphocytes activation, causing vasculitis
b. Acute cellular rejection: it is due to T cell infiltration
and activation (CD4 and CD8) ,type IV Hs
3) Chronic allograft failure (rejection)
Associated with Proliferation of transplant vascular
smooth muscle, interstitial fibrosis and scaring
Occur > 30 days
Due to immune and non-immune mechanisms
(hypertension, hyperlipidaemia and chronic drug toxicity)
Rx: decrease drug toxicity, control HT and
hyperlipidaemia
Investigations
HLA typing
Anti HLA Abs screenings
Donor-recipient cross matching
C4d staining that deposit in graft capillaries that useful in
early diagnosis of vascular rejection
Immunosuppressive drugs
1) Anti- prolifrative agents(azathioprine):
Inhibit DNA synthesis and block lymphocytes
proliferations
s.e. increased infection, leucopenia, hepatotoxicity
2) Calcineurin inhibitors(Ciclosporin):
Inhibit T cell signaling and block lymphocytes
proliferations
s.e. increased infection,nephrotoxicity ,HT
3) corticosteroids: decrease phagocytosis, decrease
lymphocytes activation and proliferation, decrease
cytokine production
s.e. increased infection
4) Anti T cell Abs (CD3)
Deplete T cells s.e. increased infection
Complications of immunosuppressant drugs
1) Infection with opportunistic pathogens
CMV---- Rx by ganciclovir
Pneumocystis----- Rx by trimethoprim
2)
Malignancy
Increased with virus associated tumor
EBV---- lymphoma
Human herpes v 8----Kaposi’s sarcoma
Increased lung, breast and colon cancer
Living organ donation
Shortage of organ donors is a problem
Cadaver ( RTA, brain- stem death)
Their relatives
The sale of organs is illegal

36
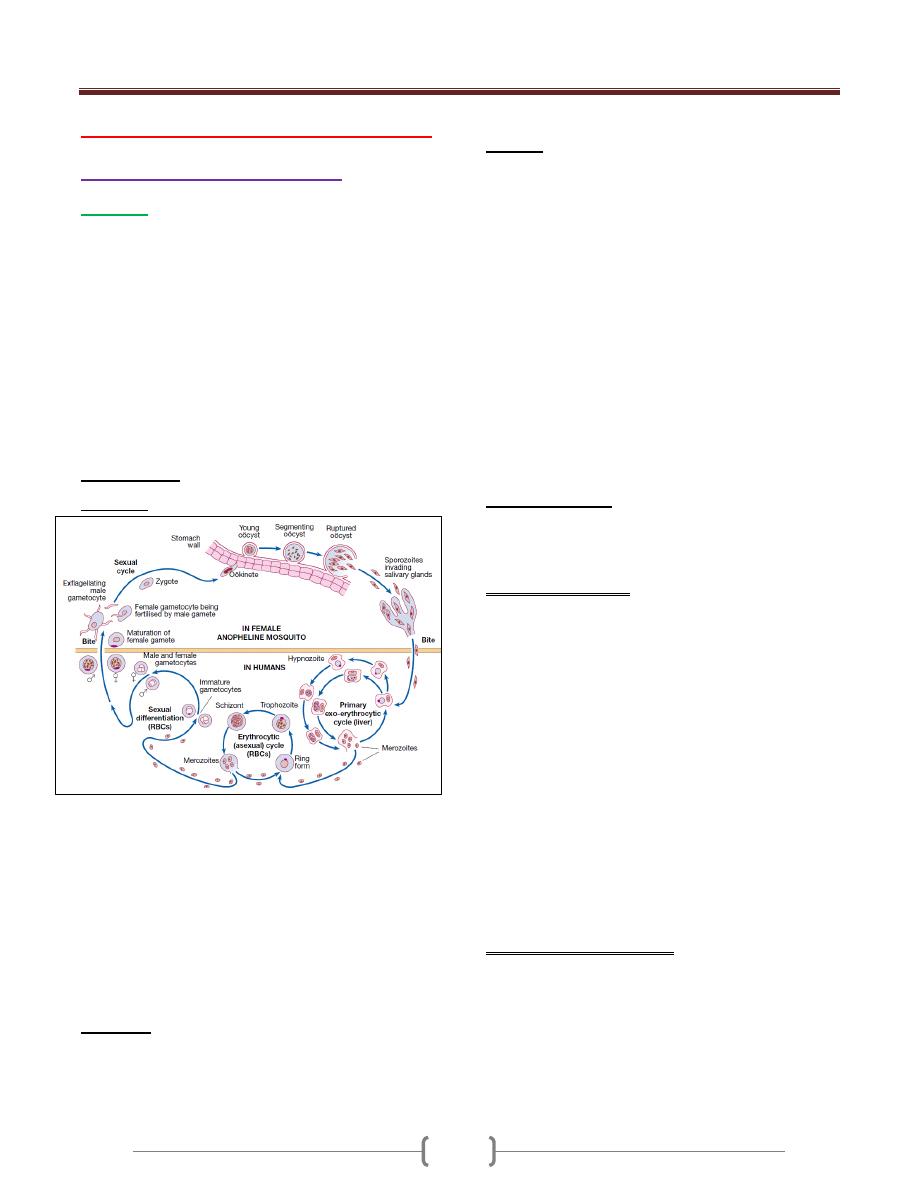
Unit 4 - Infectious disease
37
Lecture 1+2+3 - Protozoal Infections
Systemic protozoal infections
Malaria
Malaria in humans is caused by Plasmodium falciparum,
P. vivax, P. ovale, P. malariae .
It is transmitted by the bite of female anopheline
mosquitoes and occurs throughout the tropics and
subtropics at altitudes below 1500 metres. Recent
estimates have put an episodes of clinical malaria at 515
million cases per year, with two-thirds of these occurring
in sub-Saharan Africa, especially amongst children and
pregnant women. P. falciparum has now become resistant
to chloroquine and sulfadoxine-pyrimethamine, initially in
South-east Asia and now throughout Africa.
Pathogenesis
Life cycle
P. vivax and P. ovale may persist in liver cells as dormant
forms, hypnozoites, capable of developing into merozoites
months or years later. Thus the first attack of clinical
malaria may occur long after the patient has left the endemic
area & the disease may relapse after treatment if drugs that
kill only the erythrocytic stage of the parasite are given.
P. falciparum and P. malariae have no persistent exo-
erythrocytic phase but recrudescence of fever may result
from multiplication of parasites in red cells which have
not been eliminated by treatment and immune processes.
Pathology
Red cells infected with malaria are prone to hemolysis.
This is most severe with P. falciparum, which invades red
cells of all ages but especially young cells.
P. vivax and P. ovale invade reticulocytes.
P. malariae normoblasts, so that infections remain lighter.
Anaemia may be profound and is worsened by dys-
erythropoiesis, splenomegaly and depletion of folate stores.
In P. falciparum malaria, red cells containing trophozoites
adhere to vascular endothelium in post-capillary venules
in brain, kidney, liver, lungs and gut. The vessels become
congested, resulting in widespread organ damage which is
exacerbated by rupture of schizonts, liberating toxic and
antigenic substances. P. falciparum has influenced human
evolution, with the appearance of protective mutations
such as sickle-cell, thalassaemia, G6PD deficiency and
HLA-B53. P. falciparum does not grow well in red cells
that contain haemoglobin F, C or especially S.
Haemoglobin S heterozygotes (AS) are protected against
the lethal complications of malaria.
P. vivax cannot enter red cells that lack the Duffy blood
group; therefore many West Africans and African-
Americans are protected.
Clinical features
The clinical features of malaria are non-specific and the
diagnosis must be suspected in anyone returning from an
endemic area that has features of infection.
P. falciparum infection: This is the most dangerous of the
malarias and patients are either ‘killed or cured’. The
onset is often insidious, with malaise, headache and
vomiting. Cough and mild diarrhea are also common. The
fever has no particular pattern. Jaundice is common due to
haemolysis and hepatic dysfunction. The liver and spleen
enlarge and may become tender. Anaemia develops
rapidly, as does thrombocytopenia. A patient with
falciparum malaria, apparently not seriously ill, may
rapidly develop dangerous complications.
Cerebral malaria is manifested by confusion, seizures or
coma, usually without localizing signs. Children die rapidly
without any special symptoms other than fever. Immunity
is impaired in pregnancy & the parasite can preferentially
bind to a placental protein known as chondroitin sulphate A.
Abortion and intrauterine growth retardation from
parasitisation of the maternal side of the placenta are frequent
Previous splenectomy increases the risk of severe malaria.
P. vivax and P. ovale infection
In many cases the illness starts with several days of
continued fever before the development of classical bouts
of fever on alternate days.
Fever starts with a rigor.
The patient feels cold & the temperature rises to about 40 °C.
After half an hour to an hour the hot or flush phase begins.
It lasts several hours and gives way to 3- 3- profuse
perspiration and a gradual fall in temperature. The cycle is

Unit 4 - Infectious disease
38
repeated 48 hours later. Gradually the spleen and liver
enlarge and may become tender. Anaemia develops slowly.
Relapses are frequent in the first 2 years after leaving the
malarious area & infection may be acquired from blood
transfusion.
P. malariae infection
This is usually associated with mild symptoms and bouts
of fever every third day. Parasitaemia may persist for
many years with the occasional recrudescence of fever, or
without producing any symptoms. Chronic P. malariae
infection causes glomerulonephritis and longterm
nephrotic syndrome in children.
Investigations
1) Giemsa-stained thick and thin blood films should be
examined whenever malaria is suspected.
In the thick film erythrocytes are lysed, releasing all
blood stages of the parasite. This, as well as the fact that
more blood is used in thick films, facilitates the diagnosis
of low level parasitaemia. A thin film is essential to
confirm the diagnosis, to identify the species of parasite
and, in P. falciparum infections, to quantify the parasite
load (by counting the percentage of infected erythrocytes).
P. falciparum parasites may be very scanty, especially in
patients who have been partially treated. With P.
falciparum, only ring forms are normally seen in the early
stages (see Fig. 13.31); with the other species all stages of
the erythrocytic cycle may be found.
Gametocytes appear after about 2 weeks, persist after
treatment and are harmless, except that they are the source
by which more mosquitoes become infected.
2) Immunochromatographic tests for malaria antigens,
such as OptiMal® (which detects the Plasmodium lactate
dehydrogenase of several species) and ParasightF®
(which detects the P. falciparum histidine-rich protein 2),
are extremely sensitive and specific for falciparum.
malaria but less so for other species. They should be used
in parallel with blood film examination but are especially
useful where the microscopist is less experienced in
examining blood films.
3) DNA detection (PCR) is used mainly in research and is
useful for determining whether a patient has a
recrudescence of the same malaria parasite or a
reinfection with a new parasite.
Management
Mild P. falciparum malaria
Since P. falciparum is now resistant to chloroquine and
sulfadoxine- pyrimethamine (Fansidar) almost worldwide,
an artemisinin-based treatment is recommended. Co-
artemether (CoArtem® or Riamet®) contains artemether
and lumefantrine and is given as 4 tablets at 0, 8, 24, 36,
48 and 60 hours. Alternatives are quinine by mouth (600
mg of quinine salt every 8 hours for 5–7 days), together
with or followed by either doxycycline (200 mg once
daily for 7 days) or clindamycin (450 mg every 8 hours
for 7 days) or atovaquone-proguanil (Malarone®, 4
tablets once daily for 3 days).
Doxycycline & artemether should be avoided in pregnancy.
Complicated P. falciparum malaria
Severe malaria should be considered as a medical emergency.
Management includes
1) early and appropriate antimalarial chemotherapy,
2) active treatment of complications,
3) Correction of fluid, electrolyte and acid–base balance,
and avoidance of harmful ancillary treatments.
4) The treatment of choice is intravenous artesunate , as
soon as the patient has recovered sufficiently to
swallow tablets, oral artesunate
5) Quinine salt can also be used.
Management of non-falciparum malaria
P. vivax, P. ovale and P. malariae infections should be
treated with oral chloroquine: 600 mg chloroquine base
followed by 300 mg base in 6 hours, then 150 mg base
12- hourly for 2 more days. ‘radical cure’ is now achieved
in most patients with P. vivax or P. ovale malaria using a
course of primaquine (15 mg daily for 14 days), which
destroys the hypnozoite phase in the liver.
Note:
Development of a fully protective malaria vaccine
is still some way off, which is not surprising considering
that natural immunity, is incomplete and not long-lived.
There is, however, some evidence that vaccination can
reduce the incidence of severe malaria in populations.
Trial vaccines are being evaluated in Africa.
African trypanosomiasis (sleeping sickness)
African sleeping sickness is caused by trypanosomes
conveyed to humans by the bites of infected tsetse flies,
and is unique to sub-Saharan Africa.
Trypanosoma brucei gambiense trypanosomiasis has a
wide distribution in West and Central Africa.
T. brucei rhodesiense trypanosomiasis is found in parts of
East and Central Africa, where it is currently on the increase.
Clinical features
A bite by a tsetse fly is painful and commonly becomes
inflamed, but if trypanosomes are introduced, the site may

Unit 4 - Infectious disease
39
again become painful and swollen about 10 days later
(‘trypanosomal chancre’) and the regional lymph nodes
enlarge (‘Winterbottom’s sign’). Within 2–3 weeks of
infection the trypanosomes invade the blood stream. The
disease is characterised by an early haematolymphatic
stage and a late encephalitic stage in which the parasite
crosses the blood–brain barrier and chronic
encephalopathy develops.
Rhodesiense infections
In these infections the disease is more acute and severe
than in gambiense infections, so that within days or a few
weeks the patient is usually severely ill and may have
developed pleural effusions and signs of myocarditis or
hepatitis. There may be a petechial rash. The patient may
die before there are signs of involvement of the CNS. If
the illness is less acute, drowsiness, tremors and coma
develop.
Gambiense infections
The distinction between early and late stages may not be
apparent in gambiense infections. The disease usually
runs a slow course over months or years, with irregular
bouts of fever and enlargement of lymph nodes. The
spleen and liver may become palpable. After some
months without treatment, the CNS is invaded. This is
shown clinically by headache and changed behaviour,
blunting of higher mental functions, insomnia by night
and sleepiness by day, mental confusion and eventually
tremors, pareses, wasting, coma and death.
Investigations
1) Thick & thin blood films stained will reveal trypanosomes.
2) The Concentration methods include buffy coat
microscopy & miniature anion exchange chromatography.
3) Rapid card agglutination trypanosomiasis test (CATT) for
antibody detection.
4) If the CNS is affected, CSF analysis which reveals
pleocytosis, increased protein, pressure and diminished
glucose. Presence of high level IgM in CSF is suggestive
for trypanosomiasis.
Management
Before CNS involvement, intravenous suramin, for
rhodesiense infections.
For gambiense infections, intramuscular or intravenous
pentamidine.
Once the nervous system is affected, treatment with
melarsoprol is effective for both East &West African
diseases.
American trypanosomiasis (Chagas’ disease)
The cause is Trypanosoma cruzi, transmitted to humans
from the faeces of a reduviid (triatomine) bug in which
the trypanosomes have a cycle of development before
becoming infective to humans.
There are 2 phases of the disease: acute & chronic phases.
Investigations
1) T. cruzi is easily detectable in a blood film in the acute illness
2) In chronic disease it may be recovered in up to 50% of
cases by xenodiagnosis,.
3) Parasite DNA detection by PCR in the patient’s blood is a
highly sensitive method for documentation of infection
and, in addition, can be employed in faeces of bugs used
in xenodiagnosis tests to improve sensitivity.
4) Antibody detection is also highly sensitive (99%).
Management
Parasiticidal agents are used to treat the acute phase,
congenital disease and early chronic phase (within 10
years of infection).
1) Nifurtimox is given orally.
2) Benznidazole is an alternative.
3) Specific drug treatment of the chronic form is now
increasingly favoured, but in the cardiac or digestive
‘mega’ diseases it does not reverse established tissue
damage. Surgery may be needed.
Toxoplasmosis
Toxoplasma gondii is an intracellular parasite
Transmission
1- Via oöcyst- contaminated soil, salads and vegetables.
2- Ingestion or tasting of raw or undercooked meats
containing tissue cysts. Sheep, pigs and rabbits are the
most common meat sources.
Outbreaks of toxoplasmosis have been linked to the
consumption of unfiltered water.
Clinical features
1) In most immunocompetent individuals, including children
and pregnant women, the infection goes unnoticed. In
approximately 10% of patients it causes a self-limiting
illness, most common in adults aged 25–35 years.
2) The most common presenting feature is painless
lymphadenopathy, either local or generalised. In
particular, the cervical nodes are involved, but
mediastinal, mesenteric or retroperitoneal groups may be
Unit 4 - Infectious disease
40
affected. The spleen is seldom palpable. Most patients
have no systemic symptoms, but some complain of
malaise, fever, fatigue, muscle pain, sore throat and
headache. Complete resolution usually occurs within a
few months, although symptoms and lymphadenopathy
tend to fluctuate unpredictably and some patients do not
recover completely for a year or more.
3) Very infrequently, patients may develop encephalitis,
myocarditis, polymyositis, pneumonitis or hepatitis.
4) Retinochoroiditis is nearly always the result of congenital
infection but has also been reported in acquired disease.
Congenital toxoplasmosis
Acute toxoplasmosis, mostly subclinical, affects 0.3– 1%
of pregnant women, with an approximately 60%
transmission rate to the fetus which increases with
increasing gestation.
Seropositive females infected 6 months before conception
have no risk of fetal transmission.
Congenital disease affects approximately 40% of infected
fetuses, and is more likely and more severe with infection
early in gestation. Many fetal infections are subclinical at
birth but long term sequelae include retinochoroiditis,
microcephaly and hydrocephalus.
Investigations
1) Immunocompromised patients diagnosis often requires
direct detection of parasites.
2) Serology is often used in immunocompetent individuals.
a. The Sabin–Feldman dye test (indirect fluorescent
antibody test), which detects IgG antibody. Recent
infection is indicated by a fourfold or greater increase in
titre when paired sera are tested in parallel. Peak titres of
1/1000 or more are reached within 1–2 months of the
onset of infection, and the dye test then becomes an
unreliable indicator of recent infection. The detection of
significant levels of Toxoplasma-specific IgM antibody
may be useful in confirming acute infection. False
positives or persistence of IgM antibodies for years after
infection make interpretation difficult; however, negative
IgM antibodies virtually rule out acute infection.
b. During pregnancy it is critical to differentiate between
recent & past infection; the presence of high-avidity IgG
antibodies excludes infection acquired in the preceding 3–
4 months.
3) If necessary, the presence of Toxoplasma organisms in a
lymph node biopsy can be sought by staining sections
histochemically with T. gondii antiserum, or by the use of
PCR to detect Toxoplasma-specific DNA.
Management
1) In immunocompetent subjects uncomplicated toxoplasmosis
is self-limiting and responds poorly to antimicrobial therapy.
2) Treatment with pyrimethamine, sulfadiazine and folinic
acid is therefore usually reserved for rare cases of severe
or progressive disease, and for infection in
immunocompromised patients.
3) In a pregnant woman with an established recent infection,
spiramycin (3 g daily in divided doses) should be given
until term. Once fetal infection is established, treatment
with sulfadiazine and pyrimethamine plus calcium
folinate is recommended (spiramycin does not cross the
placental barrier).
Leishmaniasis
Leishmaniasis is caused by unicellular flagellate
intracellular protozoa belonging to the genus Leishmania
(order Kinetoplastidae). There are 21 leishmanial species
which cause several diverse clinical syndromes, which
can be placed into three broad groups:
• Visceral leishmaniasis (VL, kala-azar)
• Cutaneous leishmaniasis (CL)
• Mucosal leishmaniasis (ML).
Epidemiology and transmission
Zoonotic transmission of parasites from animals (chiefly
canine and rodent reservoirs) to humans through
phlebotomine sandfly vectors, Humans are the only
known reservoir (anthroponotic) in major VL foci in India
and for transmission of leishmaniasis between injection
drug-users.
Visceral leishmaniasis (VL, kala-azar)
VL is caused by the protozoon Leishmania donovani
complex including:
1- L. donovani, 2- L. infantum. 3- L. chagasi.
India, Sudan, Bangladesh and Brazil account for 90% of
cases of VL, while other affected regions include the
Mediterranean, East Africa, China, Arabia, and other
South American countries .
In addition to sandfly transmission,
VL has also been reported to follow blood transfusion and
disease can present unexpectedly in immunosuppressed
patients—for example, after renal transplantation and in
HIV infection. The great majority of people infected
remain asymptomatic.

Unit 4 - Infectious disease
41
In visceral diseases the spleen, liver, bone marrow and
lymph nodes are primarily involved.
Clinical features
On the Indian subcontinent adults and children are equally
affected; on other continents VL is predominantly a
disease of small children and infants, except in adults with
HIV co-infection.
The incubation period ranges from weeks to months
(occasionally several years).
1) The first sign of infection is High fever, usually
accompanied by rigor and chills. Fever intensity decreases
over time and patients may become afebrile for
intervening periods ranging from weeks to months. This is
followed by a relapse of fever, often of lesser intensity.
2) Splenomegaly develops quickly in the first few weeks and
becomes massive as the disease progresses.
3) Moderate hepatomegaly occurs later.
4) Lymphadenopathy is seen in the majority of cases in
Africa, the Mediterranean and South America, but is rare
on the Indian subcontinent.
5) Blackish discoloration of the skin, from which the disease
derived its name, kala-azar (the Hindi word for ‘black
fever’), is a feature of advanced illness & is now rarely seen
6) Pancytopenia is a common feature. Moderate to severe
anaemia develops rapidly, and can result in congestive
cardiac failure and associated clinical features.
Thrombocytopenia, often compounded by hepatic
dysfunction, may result in bleeding from the retina,
gastrointestinal tract and nose.
7) In advanced illness, hypoalbuminaemia may manifest as
pedal oedema, ascites and anasarca (gross generalised
oedema and swelling).
8) As the disease advances, there is profound
immunosuppression.and secondary infections are very
common. These include tuberculosis, pneumonia, severe
amoebic or bacillary dysentery, gastroenteritis, herpes
zoster and chickenpox. Skin infections, boils, cellulitis
and scabies are common.
9) Without adequate treatment most patients with clinical
VL die.
Investigations
1) Pancytopenia is the most dominant feature, with
granulocytopenia and monocytosis.
2) Polyclonal hypergammaglobulinaemia, chiefly IgG
followed by IgM, and hypoalbuminaemia are seen later.
3) Demonstration of amastigotes (Leishman–Donovan bodies)
in splenic smears is the most efficient means of diagnosis,
with 98% sensitivity); however, it carries a risk of serious
haemorrhage in inexperienced hands. Safer methods, such
as bone marrow or lymph node smears, are not as sensitive.
Parasites may be demonstrated in buffy coat smears,
especially in immunosuppressed patients. Sensitivity can be
improved by culturing the aspirate material or by PCR for
DNA detection and species identification, but these tests
can only be performed in specialised laboratories.
4) Serodiagnosis, by ELISA or immunofluorescence
antibody test, is employed in developed countries.
5) In endemic regions, a highly sensitive and specific direct
agglutination test using stained promastigotes and rapid
immunochromatographic k39 strip test have become
popular. These tests remain positive for several months
after cure has been achieved, so do not predict response to
treatment or relapse. A significant proportion of the
healthy population in an endemic region will be positive
for these tests due to past exposure.
6) Formal gel (aldehyde) for detection of raised globulin has
limited value and should not be employed for the
diagnosis of VL.
Management
1) Pentavalent antimonials
2) Amphotericin B, The antifungal drug,
3) Other drugs
a. The oral drug miltefosine, an alkyl phospholipid, has been
approved in several countries for the treatment of VL.
b. Paromomycin is an aminoglycoside that has undergone
trials in India and Africa India for the treatment of VL.
c. Pentamidine isetionate was used to treat Sb-refractory
patients with VL.
Post-kala-azar dermal leishmaniasis (PKDL)
After treatment and apparent recovery from the visceral
disease in India and Sudan, some patients develop
dermatological manifestations due to local parasitic infection.
Clinical features
In India dermatological changes occur in a small minority
of patients 6 months to ≥3 years after the initial infection.
They are seen as macules, papules, nodules (most
frequently) and plaques which have a predilection for the
face, especially the area around the chin. The face often
appears erythematous Hypopigmented macules can occur
over all parts of the body and are highly variable in extent
and location. There are no systemic symptoms and no
spontaneous healing.

Unit 4 - Infectious disease
42
Investigations and management
The diagnosis is clinical, supported by demonstration of
scanty parasites in lesions by slit-skin smear and culture.
Immunofluorescence and immunohistochemistry may
demonstrate the parasite in skin tissues.
In the majority of patients serological tests (direct
agglutination test or k39 strip tests) are positive.
Treatment of PKDL is difficult. In India, sodium
stiobogluconate for 120 days or several courses of
amphotericin B infusions are required.
Cutaneous and mucosal leishmaniasis
Cutaneous leishmaniasis (CL)
CL (oriental sore) occurs in both the Old World and the
New World (the Americas). Transmission is by sandfly .
In the Old World, CL is mild. It is found around the
Mediterranean basin, throughout the Middle East and
Central Asia as far as Pakistan, and in sub-Saharan West
Africa and Sudan .
The causative organisms for Old World zoonotic CL are
L. major, L. tropica and L. aethiopica .
Anthroponotic CL is caused by L. tropica, and is confined
to urban or suburban areas of the Old World. Afghanistan
is currently the biggest focus, but infection is endemic in
Pakistan, the western deserts of India, Iran, Iraq, Syria and
other areas of the Middle East.
New World CL is a more significant disease, which may
disfigure the nose, ears and mouth & is causedby the L.
mexicana complex (comprising L. mexicana, L. amazonensis
& L. venezuelensis) and by the Viannia subgenus L. (V.)
brasiliensis complex (comprising L. (V.) guyanensis, L.
(V.) panamensis, L. (V.) brasiliensis and L. (V.) peruviana).
CL is commonly imported and should be considered in
the differential diagnosis of an ulcerating skin lesion,
especially in travellers who have visited endemic areas of
the Old World or forests in Central and South America.
Clinical features
The incubation period is typically 2–3 months (range 2
weeks to 5 years).
In all types of CL, the common feature is development of
a papule followed by ulceration of the skin with raised
borders, usually at the site of the bite of the vector.
Lesions, single or multiple, start as small red papules that
increase gradually in size, reaching 2–10 cm in diameter.
A crust forms, overlying an ulcer with a granular base .
These ulcers develop a few weeks or months after the
bite. There can be satellite lesions, especially in L. major
and occasionally in L. tropica infections.
Regional lymphadenopathy, pain, pruritus and secondary
bacterial infections may occur.
Clinically, lesions of L. mexicana and L. peruviana closely
resemble those seen in the Old World, but lesions on the
pinna of the ear are common & are chronic & destructive.
L. mexicana is responsible for chiclero ulcers, the self-
healing sores of Mexico.
If immunity is good, there is usually spontaneous healing
in L. tropica, L. major and L. mexicana lesions. In some
patients with anergy to Leishmania, the skin lesions of L.
aethiopica, L. mexicana and L. amazonensis infections
progress to the development of diffuse CL; this is
characterized by spread of the infection from the initial
ulcer, usually on the face, to involve the whole body in
the form of non-ulcerative nodules. Occasionally, in L.
tropica infections, sores that have apparently healed
relapse persistently (recidivans or lupoid leishmaniasis).
Mucosal leishmaniasis (ML)
The Viannia subgenus extends widely from the Amazon
basin as far as Paraguay and Costa Rica, and is
responsible for deep sores and ML.
In L. (V.) brasiliensis complex infections, cutaneous
lesions may be followed by mucosal spread of the disease
simultaneously or even years later. between 2% and 40%
of infected persons develop ‘espundia’, metastatic lesions
in the mucosa of the nose or mouth. This is characterised
by thickening and erythema of the nasal mucosa, typically
starting at the junction of the nose and upper lip. Later,
ulceration develops. The lips, soft palate, fauces and
larynx may also be invaded and destroyed, leading to
considerable suffering and deformity.
There is no spontaneous healing, and death may result
from severe respiratory tract infections due to massive
destruction of the pharynx.
Investigations in CL and ML
1) CL is often diagnosed on the basis of clinical
characteristics of the lesions
2) Parasitological confirmation to detect Amastigotes on a
slit-skin smear with Giemsa staining; is also important to
exclude other disease.
3) Cultured from the sores early during the infection.
Management of CL and ML
1) Small lesion: may a- self-heal or treated by b- freezing
with liquid nitrogen or curettage.
2) In CL, topical application of paromomycin 15% plus
methylbenzethonium chloride 12% is beneficial.
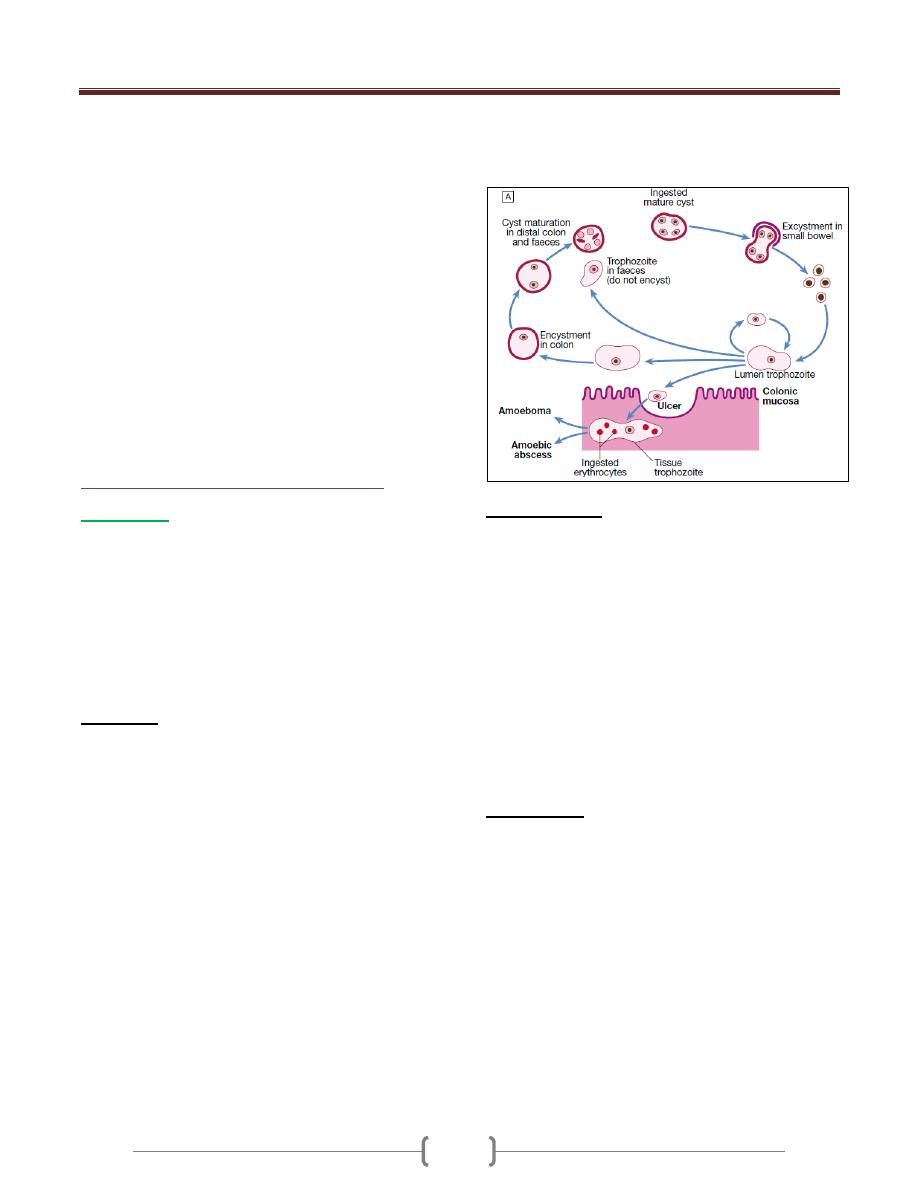
Unit 4 - Infectious disease
43
3) Intralesional antimony (Sb: 0.2–0.8 mL/lesion) up to 2 g
seems to be rapidly effective in suitable cases, well
tolerated and economic, and is safe in patients with
cardiac, liver or renal diseases.
4) In ML, and in( CL when the lesions are multiple or in a
disfiguring site),
a- parenteral Sb in a dose of 20 mg/kg/day (usually given
for 20 days for CL and 28 days for ML),
b- Conventional or liposomal amphotericin B (see
treatment of VL above).
5) Two to four doses (2–4 mg/kg) of alternate-day
administration of pentamidine are effective in New World
CL. In ML, 8 injections of pentamidine (4 mg/kg on
alternate days) cure the majority of patients.
6) Ketoconazole 600 mg daily for 4 weekshas shown some
potential against L. mexicana infection.
Gastrointestinal protozoal infections
Amoebiasis
Amoebiasis is caused by Entamoeba histolytica, which is
spread between humans by its cysts.
Two non-pathogenic Entamoeba species (E. dispar and E.
moshkovskii) are morphologically identical to E.
histolytica, and are distinguishable only by molecular
techniques, isoenzyme studies or monoclonal antibody
typing. However, only E. histolytica causes amoebic
dysentery or liver abscess.
Pathology
Cysts of E. histolytica are ingested in water or uncooked
foods contaminated by human faeces.
In the colon, vegetative trophozoite forms emerge from
the cysts. The parasite may invade the mucous membrane
of the large bowel, producing lesions that are maximal in
the caecum but found as far down as the anal canal. These
are flask-shaped ulcers, varying greatly in size and
surrounded by healthy mucosa. A localised granuloma
(amoeboma), presenting as a palpable mass in the rectum
or a filling defect in the colon on radiography, is a rare
complication which should be differentiated from colonic
carcinoma. Amoebic ulcers may cause severe
haemorrhage but rarely perforate the bowel wall.
Amoebic trophozoites can emerge from the vegetative
cyst from the bowel and be carried to the liver in a portal
venule. They can multiply rapidly and destroy the liver
parenchyma, causing an abscess.
The liquid contents at first have a characteristic pinkish
colour which may later change to chocolate brown (like
anchovy sauce).
Cutaneous amoebiasis, though rare, causes progressive
genital, perianal or peri-abdominal surgical wound ulceration.
Clinical features
Intestinal amoebiasis—amoebic dysentery
1) Most amoebic infections are asymptomatic.
2) The incubation period of amoebiasis ranges from 2 weeks
to many years, followed by a chronic course with
abdominal pains and two or more unformed stools a day.
3) Offensive diarrhoea alternating with constipation, and
blood or mucus in the stool, are common.
4) There may be abdominal pain, especially right lower
quadrant (which may simulate acute appendicitis).
5) A dysenteric presentation with passage of blood,
simulating bacillary dysentery or ulcerative colitis, occurs
particularly in older people, in the puerperium and with
superadded pyogenic infection of the ulcers.
Investigations
1) The stool & any exudate should be examined at once under
the microscope for motile trophozoites containing RBCs.
Movements cease rapidly as the stool preparation cools.
2) Several stools may need to be examined in chronic
amoebiasis before cysts are found. Sigmoidoscopy may
reveal typical flask-shaped ulcers, which should be
scraped and examined immediately for E. histolytica.
3) In endemic areas one-third of the population are
symptomless passers of amoebic cysts.
4) Serum antibodies are detectable by immunofluorescence in
over 95% of patients with hepatic amoebiasis & intestinal
amoeboma, but in only about 60% of dysenteric amoebiasis
5) DNA detection by PCR shown to be useful in diagnosis of
E. histolytica infections but is not generally available.

Unit 4 - Infectious disease
44
Amoebic liver abscess
1) The abscess is usually found in the right hepatic lobe.
2) There may not be associated diarrhoea. Early symptoms
may be local discomfort only and malaise; later, a
swinging temperature and sweating may develop, usually
without marked systemic symptoms or associated
cardiovascular signs.
3) An enlarged, tender liver, cough and pain in the right
shoulder are characteristic, but symptoms may remain
vague and signs minimal.
4) A large abscess may penetrate the diaphragm and rupture
into the lung, from where its contents may be coughed up.
5) Rupture into the pleural cavity, the peritoneal cavity or
pericardial sac is less common but more serious.
Investigations
1) An amoebic abscess of the liver is suspected on clinical
grounds; there is often a neutrophil leucocytosis and a
raised right hemidiaphragm on chest X-ray.
2) Confirmation is by ultrasonic scanning.
3) Aspirated pus from an amoebic abscess has the
characteristic anchovy sauce or chocolate brown
appearance but only rarely contains free amoebae .
Management
1) Intestinal and early hepatic amoebiasis responds quickly
to oral metronidazole (800 mg 8-hourly for 5–10 days) or
other long-acting nitroimidazoles like tinidazole or
ornidazole (both in doses of 2 g daily for 3 days).
Nitaxozanide (500 mg 12-hourly for 3 days) is an
alternative drug.
2) Either diloxanide furoate or paromomycin, in doses of
500 mg orally 8-hourly for 10 days after treatment, should
be given to eliminate luminal cysts.
3) If a liver abscess is large or threatens to burst, or if the
response to chemotherapy is not prompt, aspiration is
required and is repeated if necessary.
Rupture of an abscess into the pleural cavity, pericardial
sac or peritoneal cavity necessitates immediate aspiration
or surgical drainage. Small serous effusions resolve
without drainage.
Giardiasis
Infection with Giardia lamblia is found world-wide and is
common in the tropics.
In cystic form it remains viable in water for up to 3 months
and infection usually occurs by ingesting contaminated
water. Its flagellar trophozoite form attaches to the
duodenal and jejunal mucosa, causing inflammation.
Incubation period of 1–3 weeks,
Clinical features
There is diarrhoea, abdominal pain, weakness, anorexia,
nausea and vomiting.
Investigations
1) On examination there may be abdominal distension and
tenderness. Stools obtained at 2–3-day intervals should be
examined for cysts.
2) Duodenal or jejunal aspiration by endoscopy gives a
higher diagnostic yield.
3) The ‘string test’ may be used, in which one end of a piece
of string is passed into the duodenum by swallowing,
retrieved after an overnight fast, and expressed fluid
examined for the presence of G. lamblia trophozoites.
4) A number of stool antigen detection tests are available.
Jejunal biopsy specimens may show G. lamblia on the
epithelial surface.
Management
Treatment is with a single dose of tinidazole 2 g, or
metronidazole 400 mg 8-hourly for 10 days.
Cryptosporidiosis
Cryptosporidium spp. are coccidian protozoal parasites of
humans and domestic animals.
Infection is acquired by the faecal–oral route through
contaminated water supplies.
The incubation period is approximately 7–10 days, and
is followed by watery diarrhoea and abdominal cramps.
The illness is usually self-limiting, but in
immunocompromised patients, especially those with HIV,
the illness can be devastating, with persistent severe
diarrhea and substantial weight loss.
Cyclosporiasis
Cyclospora cayetanensis is a globally distributed
coccidian protozoal parasite of humans.
Infection is acquired by ingestion of contaminated water.
The incubation period is approximately 2–11 days, and
is followed by acute onset of diarrhoea with abdominal
cramps, which may remit and relapse.
Although usually self-limiting, the illness may last as long
as 6 weeks with significant associated weight loss and
malabsorption, and is more severe in
immunocompromised individuals.
Diagnosis is by detection of oöcysts on faecal
microscopy.
Treatment may be necessary in a few cases & the agent
of choice is co-trimoxazole 960 mg 12-hourly for 7 days.
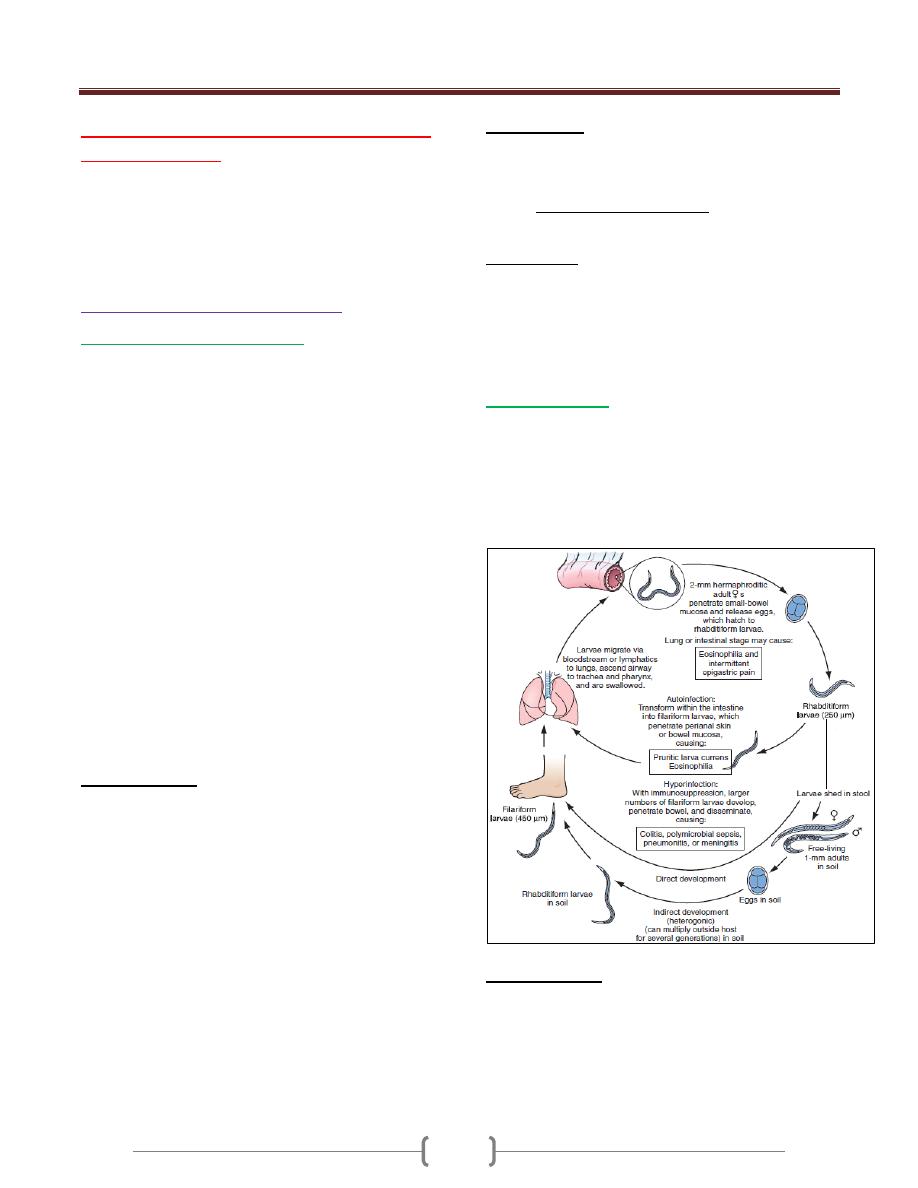
Unit 4 - Infectious disease
45
Lecture 4+5+6+7 - Infections caused
by Helminthes
Helminths (from the Greek Helmins, meaning worm) are
large multicellular organisms with complex tissues & organs
They include three groups of parasitic worm, these are:
1- Nematodes or roundworms 2- Trematodes or flukes
3- Cestodes or tapeworms
A) Intestinal human nematodes
Ancylostomiasis (hookworm)
Ancylostomiasis is caused by parasitisation with
Ancylostoma duodenale or Necator americanus.
The adult hookworm is 1 cm long and lives in the
duodenum and upper jejunum attach themselves to the
mucosa of the small intestine by their buccal capsule.
Eggs are passed in the faeces. In warm, moist, shady soil
the larvae develop into rhabditiform and then the infective
filariform stages; they then penetrate human skin and are
carried to the lungs. After entering the alveoli they ascend
the bronchi, are swallowed and mature in the small
intestine, reaching maturity 4–7 weeks after infection. The
worms attach themselves to the mucosa of the small
intestine by their buccal capsule and withdraw blood.
The mean daily loss of blood from one A. duodenale is
0.15 mL and from N. americanus 0.03 mL.
It is one of the main causes of anaemia in the tropics &
subtropics. A. duodenale is endemic in the Far East &
Mediterranean coastal regions & is also present in Africa,
while N. americanus is endemic in West, East & Central
Africa & Central & South America, as well as in the Far East.
Clinical features
1) An allergic dermatitis, usually on the feet (ground itch),
may be experienced at the time of infection.
2) The passage of the larvae through the lungs in a heavy
infection causes a paroxysmal cough with blood-stained
sputum, associated with patchy pulmonary consolidation
and eosinophilia.
3) When the worms have reached the small intestine,
vomiting and epigastric pain resembling peptic ulcer
disease may occur.
4) Sometimes frequent loose stools are passed.
5) The degree of iron and protein deficiency which develops
depends not only on the load of worms but also on the
nutrition of the patient and especially on the iron stores.
Anaemia with high-output cardiac failure may result.
6) The mental and physical development of children may be
retarded in severe infection.
Investigations
1) There is eosinophilia.
2) The characteristic ovum can be recognised in the stool.
3) If hookworms are present in numbers sufficient to cause
anaemia, faecal occult blood testing will be positive and
many ova will be present.
Management
1) A single dose of albendazole (400 mg) is treatment of choice.
2) Alternatively, mebendazole 100 mg 12-hourly for 3 days
may be used. 3) Anaemia and heart failure associated
with hookworm infection respond well to oral iron, even
when severe; blood transfusion is rarely required.
Strongyloidiasis
Strongyloides stercoralis is a very small nematode (2 mm
× 0.4 mm) which parasitises the mucosa of the upper part
of the small intestine, often in large numbers, causing
persistent eosinophilia.
Strongyloidiasis occurs in the tropics and subtropics, and
is especially prevalent in the Far East.
Clinical features
The classic triad of symptoms consists of abdominal pain,
diarrhoea and urticaria.
Cutaneous manifestations, either urticaria or larva currens
(a highly characteristic pruritic, elevated, erythematous
lesion advancing along the course of larval migration), are
characteristic and occur in 66% of patients.

Unit 4 - Infectious disease
46
Systemic strongyloidiasis (the Strongyloides hyper-
infection syndrome), with dissemination of larvae
throughout the body occurs in association with immune
suppression (intercurrent disease, HIV& HTLV-1 infection,
corticosteroid treatment). Patients present with severe,
generalised abdominal pain, abdominal distension &shock.
Massive larval invasion of the lungs causes cough, wheeze
and dyspnoea; cerebral involvement has manifestations
ranging from subtle neurological signs to coma. Gram-
negative sepsis frequently complicates the picture.
Investigations
1) There is eosinophilia.
2) Serology (ELISA) is helpful, but definitive diagnosis
depends upon finding the larvae.
3) The faeces should be examined microscopically for motile
larvae; excretion is intermittent so repeated examinations
may be necessary.
4) Larvae can also be found in jejunal aspirate or detected
using the string test.
5) Larvae may also be cultured from faeces.
Management
1) Ivermectin 200 µg/kg as a single dose, or two doses of
200 µg/kg on successive days, is effective.
2) Alternatively, albendazole is given orally in a dose of 15
mg/kg body weight 12-hourly for 3 days. A second course
may be required.
3) For the Strongyloides hyperinfection syndrome,
ivermectin is given at 200 µg/kg on days 1, 2, 15 and 16.
Ascaris lumbricoides (roundworm)
This pale yellow nematode is 20–35 cm long.
Humans are infected by eating food contaminated with
mature ova.
Ascaris larvae hatch in the duodenum, migrate through
the lungs, ascend the bronchial tree, are swallowed and
mature in the small intestine. This tissue migration can
provoke both local and general hypersensitivity reactions,
with pneumonitis, eosinophilic granulomas, bronchial
asthma and urticaria.
Clinical features
1) Intestinal ascariasis causes symptoms ranging from
occasional vague abdominal pain through to malnutrition.
2) The large size of the adult worm & its tendency to
aggregate & migrate can result in obstructive complications
3) Tropical and subtropical areas are endemic for ascariasis,
and in these areas it causes up to 35% of all intestinal
obstructions, most commonly in the terminal ileum.
4) Obstruction can be complicated further by
intussusceptions, volvulus, haemorrhagic infarction and
perforation.
5) Other complications include blockage of the bile or
pancreatic duct and obstruction of the appendix by adult
worms.
Investigations
1) The diagnosis is made microscopically by finding ova in
the faeces.
2) Adult worms are frequently expelled rectally or orally.
3) Occasionally, the worms are demonstrated
radiographically by a barium examination.
4) There is eosinophilia.
Management
A single dose of albendazole (400 mg), pyrantel pamoate
(11 mg/kg; maximum 1 g), piperazine (4 g) or
mebendazole (100 mg 12-hourly for 3 days) is effective
for intestinal ascariasis.
Patients should be warned that they might expel numerous
whole, large worms.
Obstruction due to ascariasis should be treated with
nasogastric suction, piperazine and intravenous fluids.
Prevention
Community chemotherapy programmes have been used to
reduce Ascaris infection.
The whole community can be treated every 3 months for
several years.
Schoolchildren can be targeted; treating them lowers the
prevalence of ascariasis in the community.
Enterobius vermicularis (threadworm)
This helminth is common throughout the world. It affects
mainly children. After the ova are swallowed,
development takes place in the small intestine, but the
adult worms are found chiefly in the colon.
Clinical features
The gravid female worm lays ova around the anus,
causing intense itching, especially at night. The ova are
often carried to the mouth on the fingers and so
reinfection or human-to-human infection takes place
In females the genitalia may be involved. The adult
worms may be seen moving on the buttocks or in the stool
Investigations
Ova are detected by applying the adhesive surface of
cellophane tape to the perianal skin in the morning. This
is then examined on a glass slide under the microscope.

Unit 4 - Infectious disease
47
Management
A single dose of mebendazole 100 mg, albendazole 400 mg
,pyrantel pamoate (11 mg/kg) or piperazine (4 g) is given &
may be repeated after 2 weeks to control auto-reinfection.
If infection recurs in a family, each member should be
treated as above. During this period all nightclothes and
bed linen are laundered. Fingernails must be kept short
and hands washed carefully before meals. Subsequent
therapy is reserved for those family members who
develop recurrent infection.
Trichuris trichiura (whipworm)
Infections with whipworm are common all over the world
under unhygienic conditions.
Infection is contracted by the ingestion of earth or food
contaminated with ova which have become infective after
lying for 3 weeks or more in moist soil.
The adult worm is 3–5 cm long and has a coiled anterior
end resembling a whip.
Whipworms inhabit the caecum, lower ileum, appendix,
colon and anal canal.
There are usually no symptoms, but intense infections in
children may cause persistent diarrhoea or rectal prolapse,
and growth retardation.
Diagnosis is readily made by identifying ova in faeces.
Treatment mebendazole in doses of 100 mg 12-hourly
for 3–5 days or a single dose of albendazole 400 mg.
Tissue-dwelling human nematodes
Filarial worms are tissue-dwelling nematodes.
Disease is due to the host’s immune response to the
worms (both adult and microfilariae (larva)), particularly
dying worms.
The worms are long-lived; microfilariae survive 2–3 years
and adult worms 10–15 years. The infections are chronic
and worst in individuals constantly exposed to reinfection.
Lymphatic filariasis
Infection with the filarial worms Wuchereria bancrofti
and Brugia malayi is associated with clinical outcomes
ranging from subclinical infection to hydrocele and
elephantiasis.
The infection is widespread in tropical Africa, the North
African coast, coastal areas of Asia, Indonesia and
northern Australia, the South Pacific islands, the West
Indies and also in North and South America.
Pathology
Several factors contribute to the pathogenesis of the disease.
1) Toxins released by the adult worm cause
lymphangiectasia; this dilatation of the lymphatic vessels
leads to lymphatic dysfunction and the chronic clinical
manifestations of lymphatic filariasis, lymphoedema and
hydrocele.
2) Death of the adult worm results in acute filarial
lymphangitis.Lymphatic obstruction persists after death of
the adult worm.
3) Secondary bacterial infections cause tissue destruction.
4) The host response to microfilariae is central to the
pathogenesis of tropical pulmonary eosinophilia.
Clinical features
Acute filarial lymphangitis presents with fever, pain,
tenderness and erythema along the course of inflamed
lymphatic vessels.Inflammation of the spermatic cord,
epididymis and testis is common.
The whole episode lasts a few days but may recur several
times a year. Temporary oedema becomes more persistent
and regional lymph nodes enlarge.
Progressive enlargement, coarsening, corrugation, fissuring
and bacterial infection of the skin and subcutaneous tissue
develop gradually, causing irreversible ‘elephantiasis’.
Tropical pulmonary eosinophilia
It is a complication seen mainly in India and is likely to be
due to microfilariae trapped in the pulmonary capillaries
and destroyed by allergic inflammation.
Patients present with paroxysmal cough, wheeze & fever.
If untreated, this progresses to debilitating chronic
interstitial lung disease.
Investigations of lymphatic filariasis
1) In the earliest stages of lymphangitis the diagnosis is
made on clinical grounds, supported by eosinophilia and
sometimes by positive filarial serology.
2) Microfilariae can be found in the peripheral blood at
night, and either are seen moving in a wet blood film or
are detected by microfiltration of a sample of lysed blood.
By the time elephantiasis develops, microfilariae become
difficult to find.
3) Movement of adult worms can be seen on scrotal ultrasound.
4) PCR-based tests for detection of W. bancrofti DNA from
blood.
Management
1) Treatment of the individual is aimed at reversing and
halting disease progression. Diethylcarbamazine (DEC)
kills microfilariae and adult worms.

Unit 4 - Infectious disease
48
2) A single dose of either ivermectin (200 µg/kg) or
albendazole (400 mg) in combination with DEC (300 mg)
also eliminates microfilariae for 1 year.
Trematodes (flukes)
Schistosomiasis
There are five species of the genus Schistosoma which
commonly cause disease in humans: S. haematobium, S.
mansoni, S. japonicum, S. mekongi and S. intercalatum.
S. haematobium was discovered by Theodor Bilharz in
Cairo in 1861 and the disease is sometimes called
bilharzia or bilharziasis.
Habitat
Adult worms live in the mesenteric veins (S. mansoni, S.
japonicum, S. mekongi, and S. intercalatum) or in the
venous plexus around the lower ends of the ureters & the
urinary bladder (S. haematobium). In these sites, they start
their sexual reproduction by releasing eggs. Once deposited
in the host, eggs may stay in the mesenteric vein, be trapped
in the intestines, escape to the intestinal lumen, and migrate
by portal blood to the liver (S. mansoni, S. japonicum).
Eggs of S. haematobium may be trapped in the intestines &
bladder and may escape to the intestinal or bladder lumen.
Life cycle
After the egg being excreted with feces or urine into fresh
water, the eggs hatch and release ciliated motile miracidia
that penetrates into the snail intermediate host. Following
asexual multiplication in the snail, the development of
cercariae, the infective forms for humans, takes 4 to 7
weeks. After leaving the snails, the cercariae can survive
in fresh water for almost 72 hours. When penetration of
the skin in the human host occurs, the cercariae lose their
tails and change into schistosomula. Schistosomula
migrate to the lungs and, in about 6 weeks, mature to
adult worms and descend to their final habitat.
Pathology
This depends on the species and the stage of infection.
Most disease is due to:
1) The passage of eggs through mucosa .
2) Granulomatous reaction to eggs deposited in tissues.
The eggs of S. haematobium pass mainly through the wall
of the bladder, but may also involve rectum, seminal
vesicles, vagina, cervix and uterine tubes. Eggs of S.
haematobium may leave the vesical plexus and be carried
directly to the lung
S. mansoni and S. japonicum eggs pass mainly through
the wall of the lower bowel or are carried to the liver and
then reach the lungs after the development of portal
hypertension and consequent portasystemic collateral
circulation. In both circumstances egg deposition in the
pulmonary vasculature, and the resultant host response,
can lead to the development of pulmonary hypertension.
Clinical features
1) During the early stages of infection there may be itching
lasting 1–2 days at the site of cercarial penetration
(swimmers itch).
2) Acute schistosomiasis (Katayama syndrome) develops
after a symptom-free period of 3–5 weeks, may present
with allergic manifestations such as urticaria, fever,
muscle aches, abdominal pain, headaches, cough and
sweating.
On examination hepatomegaly, splenomegaly,
lymphadenopathy and pneumonia may be present. These
allergic phenomena may be severe in infections with S.
mansoni and S. japonicum, but are rare with S.
haematobium. The features subside after 1–2 weeks.
Chronic schistosomiasis is due to egg deposition and
occurs months to years after infection. The symptoms and
signs depend upon the intensity of infection and the
species of infecting schistosome.
Schistosoma haematobium
As adult worms can live for 20 years or more. It is highly
endemic in Egypt and East Africa, and occurs throughout
Africa and the Middle East
1) Painless terminal haematuria is usually the first and most
common symptom.
2) Frequency of micturition due to bladder neck obstruction.
3) Later the disease may be complicated by frequent urinary
tract infections, bladder or ureteric stone formation,
hydronephrosis, and ultimately renal failure with a
contracted calcified bladder.
4) Pain is often felt in the iliac fossa or in the loin, and
radiates to the groin.
5) In several endemic areas there is a strong epidemiological
association of S. haematobium infection with squamous
cell carcinoma of the bladder. 6) Intestinal
symptoms may follow involvement of the bowel wall.
Schistosoma mansoni
S. mansoni is endemic throughout Africa, the Middle
East, Venezuela, Brazil and the Caribbean.
Characteristic symptoms begin 2 months or more after
infection.

Unit 4 - Infectious disease
49
They may be slight, no more than malaise, or consist of
abdominal pain and frequent stools which contain blood-
stained mucus.
With severe advanced disease,increased discomfort from
rectal polyps may be experienced.
The early hepatomegaly is reversible, but portal hyper-
tension may cause massive splenomegaly, fatal haema-
temesis from oesophageal varices, or progressive ascites.
Liver function is initially preserved because the pathology
is fibrotic rather than cirrhotic.
Schistosoma japonicum, S. mekongi and S.
intercalatum
The clinical features resemble those of severe infection
with S. mansoni, with added neurological features. The
small and large bowel may be affected, and hepatic
fibrosis with splenic enlargement is usual. Deposition of
eggs or worms in the CNS, especially in the brain or
spinal cord, causes symptoms in about 5% of infections,
notably epilepsy, blindness, hemiplegia or paraplegia.
Investigations
1) There is marked eosinophilia.
2) Serological tests (ELISA) are useful as screening tests but
remain positive after chemotherapeutic cure.
3) In S. haematobium infection, dipstick urine testing shows
blood and albumin.
4) The eggs can be found by microscopic examination of the
centrifuged deposit of terminal stream urine . Ultrasound
is useful for assessing the urinary tract; bladder wall
thickening, hydronephrosis and bladder calcification can
be detected.
5) Cystoscopy reveals ‘sandy’ patches, bleeding mucosa and
later distortion.
6) Sigmoidoscopy may show inflammation or bleeding.
Biopsies should be examined for ova.
Management
1) The object of specific treatment is to kill the adult
schistosomes and so stop egg-laying.
2) Praziquantel is the drug of choice for all forms of
schistosomiasis.
3) Surgery may be required to deal with residual lesions such
as ureteric stricture, small fibrotic urinary bladders, or
granulomatous masses in the brain or spinal cord.
Cestodes (Tapeworms)
Cestodes are ribbon-shaped worms which inhabit the
intestinal tract. They have no alimentary system and
absorb nutrients through the tegumental surface. The
anterior end, or scolex, has suckers for attaching to the
host. From the scolex arises a series of progressively
developing segments, the proglottides, which, when shed,
may continue to show active movements. Cross-
fertilization takes place between segments. Ova, present
in large numbers in mature proglottides, remain viable for
weeks and during this period they may be consumed by
the intermediate host. Larvae liberated from the ingested
ova pass into the tissues, forming larval cysticerci.
Tapeworms cause two distinct patterns of disease either
1-Intestinal infection 2- Systemic cysticercosis.
Taenia saginata (beef tapeworm) and Diphyllobothrium
latum (fish tapeworm) cause only intestinal infection,
following human ingestion of intermediate hosts that
contain cysticerci (the larval stage of the tapeworm).
Taenia solium causes intestinal infection if a cysticerci-
containing intermediate host is ingested, and cysticercosis
(systemic infection from larval migration) if ova are ingested.
Echinococcus granulosus (dog tapeworm) does not cause
human intestinal infection, but causes hydatid disease
(which is analogous to cysticercosis) following ingestion
of ova and subsequent larval migration.
Intestinal tapeworm
Humans acquire tapeworm by eating undercooked beef
infected with the larval stage of T. saginata, undercooked
pork containing the larval stage of T. solium, or
undercooked freshwater fish containing larvae of D.
latum. Usually only one adult tapeworm is present in the
gut but up to ten have been reported.
The ova of T. saginata and T. solium are indistinguishable
microscopically. However, examination of scolex and
proglottides can differentiate between them. T. solium has a
rostellum and two rows of hooklets on the scolex, and
discharges multiple proglottides (3–5) attached together
with lower degrees of uterine branching (approximately
10); T. saginata has only four suckers in its scolex, and
discharges single proglottids with greater uterine branching.
Taenia saginata
Infection with T. saginata occurs in all parts of the world.
The adult worm may be several meters long and produces
little or no intestinal upset in human beings, but
knowledge of its presence, by noting segments in the
faeces or on underclothing, may distress the patient.
Ova may be found in the stool.
Unit 4 - Infectious disease
50
Praziquantel is the drug of choice; niclosamide or
nitazoxanide are alternatives.
Prevention depends on efficient meat inspection and the
thorough cooking of beef.
Taenia solium
T. solium, the pork tapeworm, is common in central
Europe, South Africa, South America and parts of Asia. It
is not as large as T. saginata. The adult worm is found
only in humans following the eating of undercooked pork
containing cysticerci.
Niclosamide, followed by a mild laxative (after 1–2 hours)
to prevent retrograde intestinal autoinfection, is effective
for intestinal infection. Cooking pork well prevents
intestinal infection. Great care must be taken by nurses and
other adults while attending a patient harbouring an adult
worm to avoid ingestion of ova or segments.
Cysticercosis
Human cysticercosis is acquired by ingesting T. solium
tapeworm ova, from either contaminated fingers or food .
The larvae are liberated from eggs in the stomach,
penetrate the intestinal mucosa and are carried to many
parts of the body where they develop and form cysticerci,
0.5–1 cm cysts that contain the head of a young worm.
They do not grow further or migrate.
Common locations are the subcutaneous tissue, skeletal
muscles and brain.
Clinical features
When superficially placed, cysts can be palpated under
the skin or mucosa as pea-like ovoid bodies. Here they
cause few or no symptoms, and will eventually die and
become calcified.
Heavy brain infections, especially in children, may cause
features of encephalitis. More commonly, however,
cerebral signs do not occur until the larvae die, 5–20 years
later. Epilepsy, personality changes, staggering gait or signs
of internal hydrocephalus are the most common features.
Investigations
1) Calcified cysts in muscles can be recognised radiologically.
2) In the brain, however, less calcification takes place and
larvae are only occasionally visible by plain X-ray;
usually CT or MRI will show them.
3) Epileptic fits starting in adult life suggest the possibility
of cysticercosis if the patient has lived in or traveled to an
endemic area.
4) The subcutaneous tissue should be palpated and any
nodule excised for histology.
5) Radiological examination of the skeletal muscles may be
helpful.
6) Antibody detection is available for serodiagnosis.
Management
1) Albendazole, 15 mg/kg daily for a minimum of 8 days,
has now become the drug of choice for parenchymal
neurocysticercosis.
2) Praziquantel is another option, 50 mg/kg in three divided
doses daily for 10 days.
3) Prednisolone, 10 mg 8-hourly, is also given for 14 days,
starting 1 day before the albendazole or praziquantel.
4) In addition, anti-epileptic drugs should be given until the
reaction in the brain has subsided.
5) Operative intervention is indicated for hydrocephalus.
6) Studies from India and Peru suggest that most small
solitary cerebral cysts will resolve without treatment.
Echinococcus granulosus (Taenia echinococcus)
& hydatid disease
Dogs are the definitive hosts of the tiny tapeworm E.
granulosus. The larval stage, a hydatid cyst, normally
occurs in sheep, cattle, camels and other animals that are
infected from contaminated pastures or water. By
handling a dog or drinking contaminated water, humans
may ingest eggs. The embryo is liberated from the ovum
in the small intestine and gains access to the blood stream
and thus to the liver. The resultant cyst grows very slowly,
sometimes intermittently.
Hydatid cyst structure
It is composed of an enveloping fibrous pericyst,
laminated hyaline membrane (ectocyst) and inner
germinal layers (endocyst) which gives rise to daughter
cysts, or germinating cystic brood capsule in which larvae
(protoscolices) develop. Over time some cysts may calcify
and become non-viable.
The disease is common in the Middle East, North and
East Africa, Australia and Argentina.
E. multilocularis, which has a cycle between foxes and
voles, causes a similar but more severe infection, ‘alveolar
hydatid disease’, which invades the liver like cancer.
Clinical features
A hydatid cyst is typically acquired in childhood and may,
after growing for some years, cause pressure symptoms.
These vary, depending on the organ or tissue involved. In
nearly 75% of patients with hydatid disease the right lobe
of the liver is invaded and contains a single cyst. In others a
cyst may be found in lung, bone, brain or elsewhere.

Unit 4 - Infectious disease
51
Investigations
1) The diagnosis depends on the clinical, radiological and
ultrasound findings in a patient who has lived in close
contact with dogs in an endemic area.
2) Complement fixation and ELISA are positive in 70–90%
of patients.
Management
1) Hydatid cysts should be excised wherever possible. Great
care is taken to avoid spillage and cavities are sterilized
with 0.5% silver nitrate or 2.7% sodium chloride.
2) Albendazole (400 mg 12-hourly for 3 months) should also
be used.
3) Albendazole is now often combined with PAIR
(percutaneous puncture, aspiration, injection of scolicidal
agent and re-aspiration) to good effect.
4) Praziquantel 20 mg/kg 12-hourly for 14 days also kills
protoscolices

52

Unit 5 - Pain
53
Lecture 1 - Introduction

Unit 5 - Pain
54

Unit 5 - Pain
55
Lecture 2 - Headache

Unit 5 - Pain
56

Unit 5 - Pain
57

Unit 5 - Pain
58
Lecture 3 – Chest pain

Unit 5 - Pain
59

Unit 5 - Pain
60

Unit 5 - Pain
61

Unit 5 - Pain
62
Lecture 4 – Abdominal pain

Unit 5 - Pain
63

Unit 5 - Pain
64

Unit 5 - Pain
65

66

Unit 6 – Common diseases
67
Lecture 1 – Cough & Hemoptysis

Unit 6 – Common diseases
68

Unit 6 – Common diseases
69

Unit 6 – Common diseases
70
Lecture 2 – Breathlessness (Dyspnea) & Cyanosis

Unit 6 – Common diseases
71

Unit 6 – Common diseases
72

Unit 6 – Common diseases
73
Lecture 3 – Dysphagia

Unit 6 – Common diseases
74

Unit 6 – Common diseases
75
Lecture 4 – Hematemesis, Melena & Diarrhea

Unit 6 – Common diseases
76

Unit 6 – Common diseases
77
Lecture 5 – Constipation & Jaundice

Unit 6 – Common diseases
78

Unit 6 – Common diseases
79
Lecture 6 – Oedema & Ascites

Unit 6 – Common diseases
80

Unit 6 – Common diseases
81

82

Unit 7 - Clinical biochemistry and metabolism
83
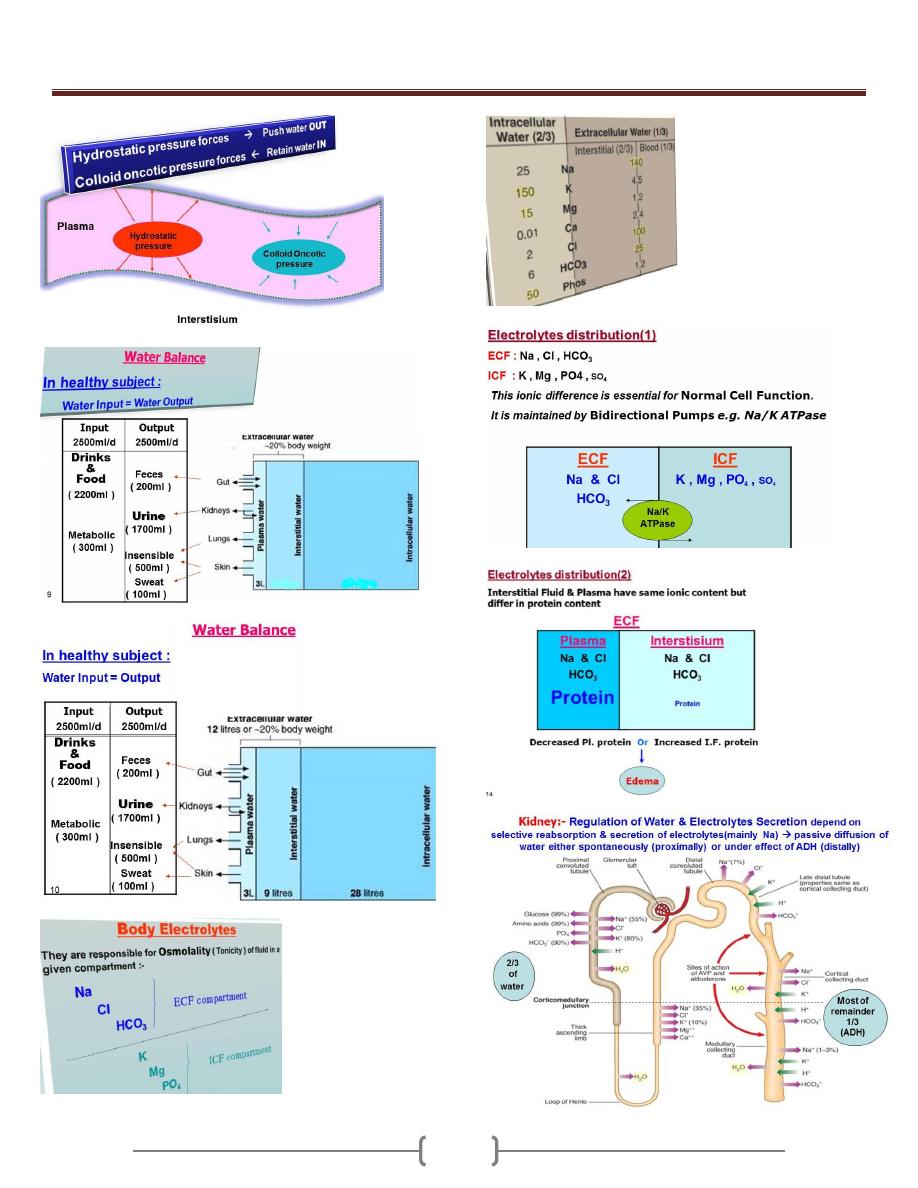
Unit 7 - Clinical biochemistry and metabolism
84

Unit 7 - Clinical biochemistry and metabolism
85
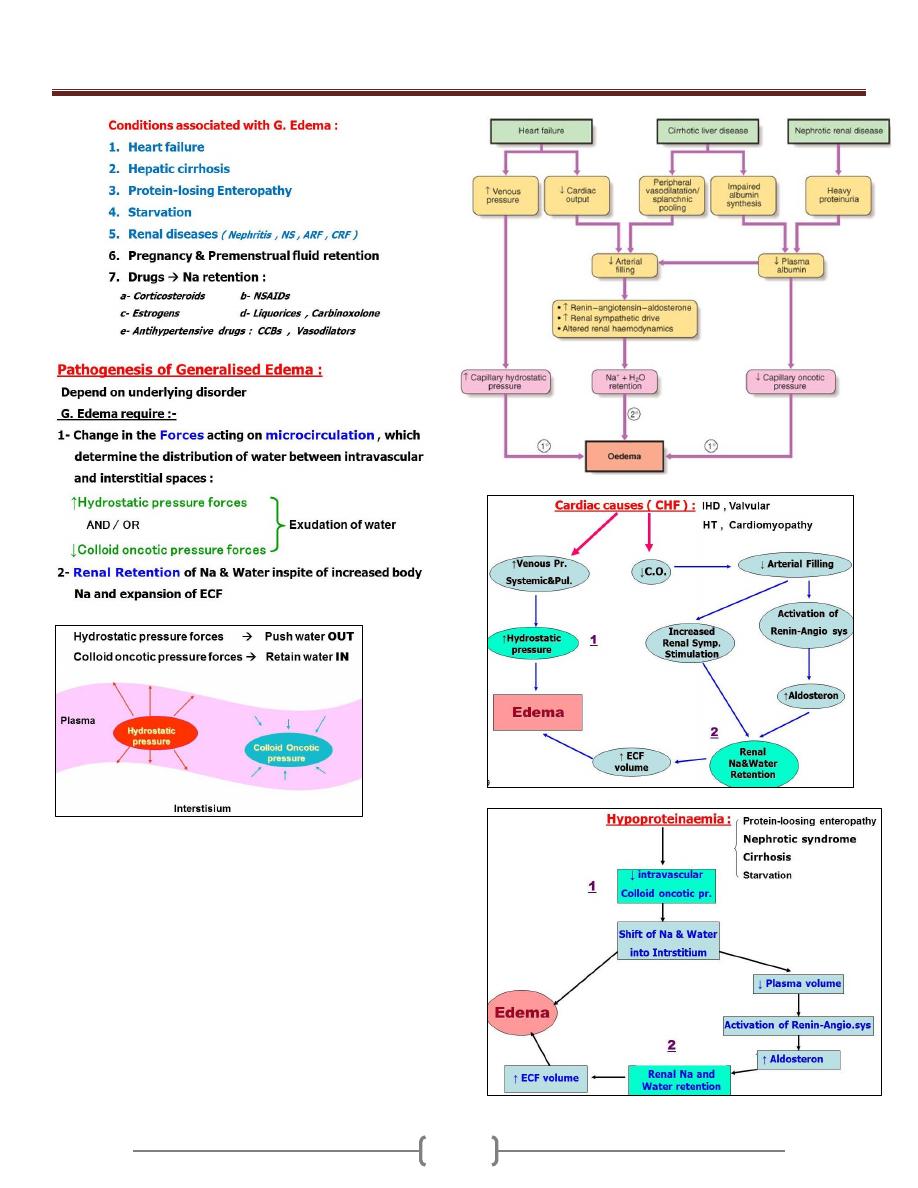
Unit 7 - Clinical biochemistry and metabolism
86
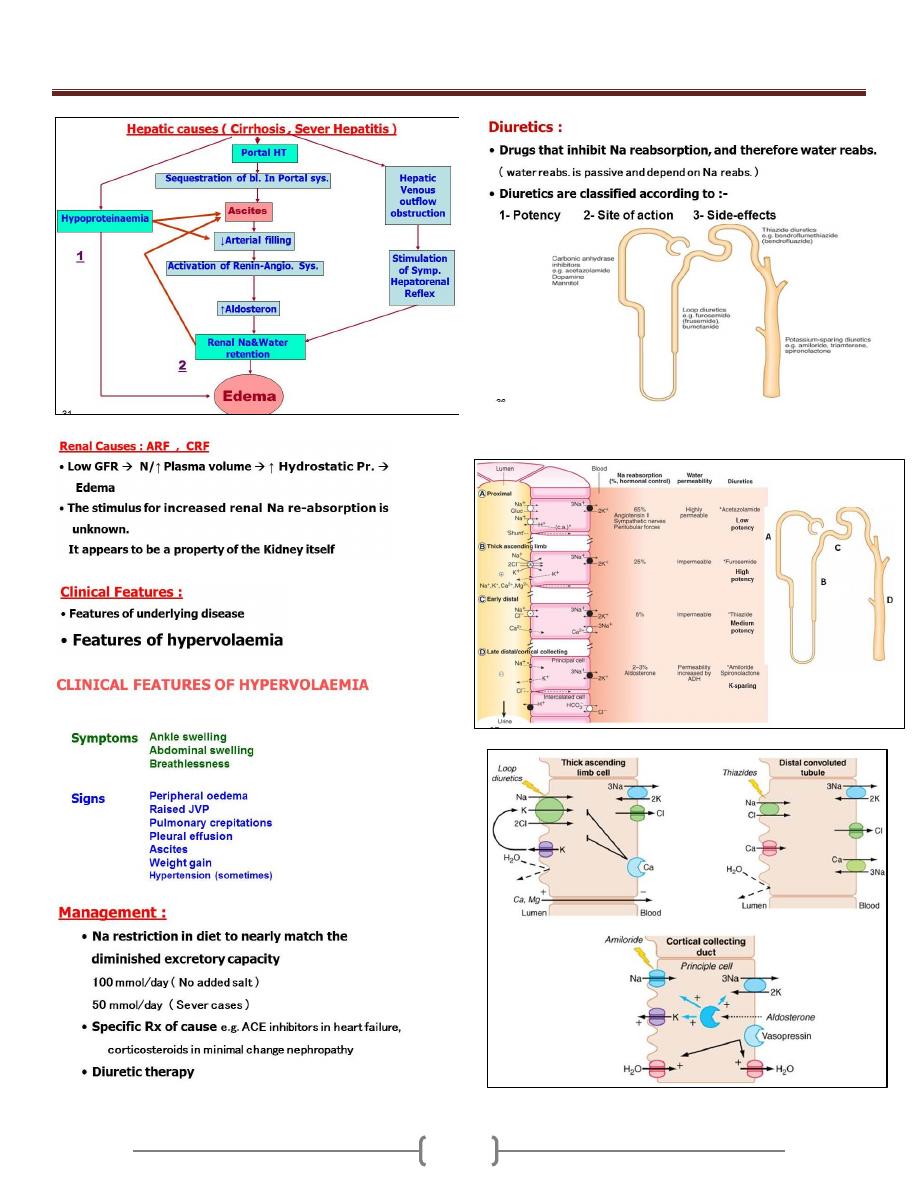
Unit 7 - Clinical biochemistry and metabolism
87

Unit 7 - Clinical biochemistry and metabolism
88

Unit 7 - Clinical biochemistry and metabolism
89

Unit 7 - Clinical biochemistry and metabolism
90

Unit 7 - Clinical biochemistry and metabolism
91
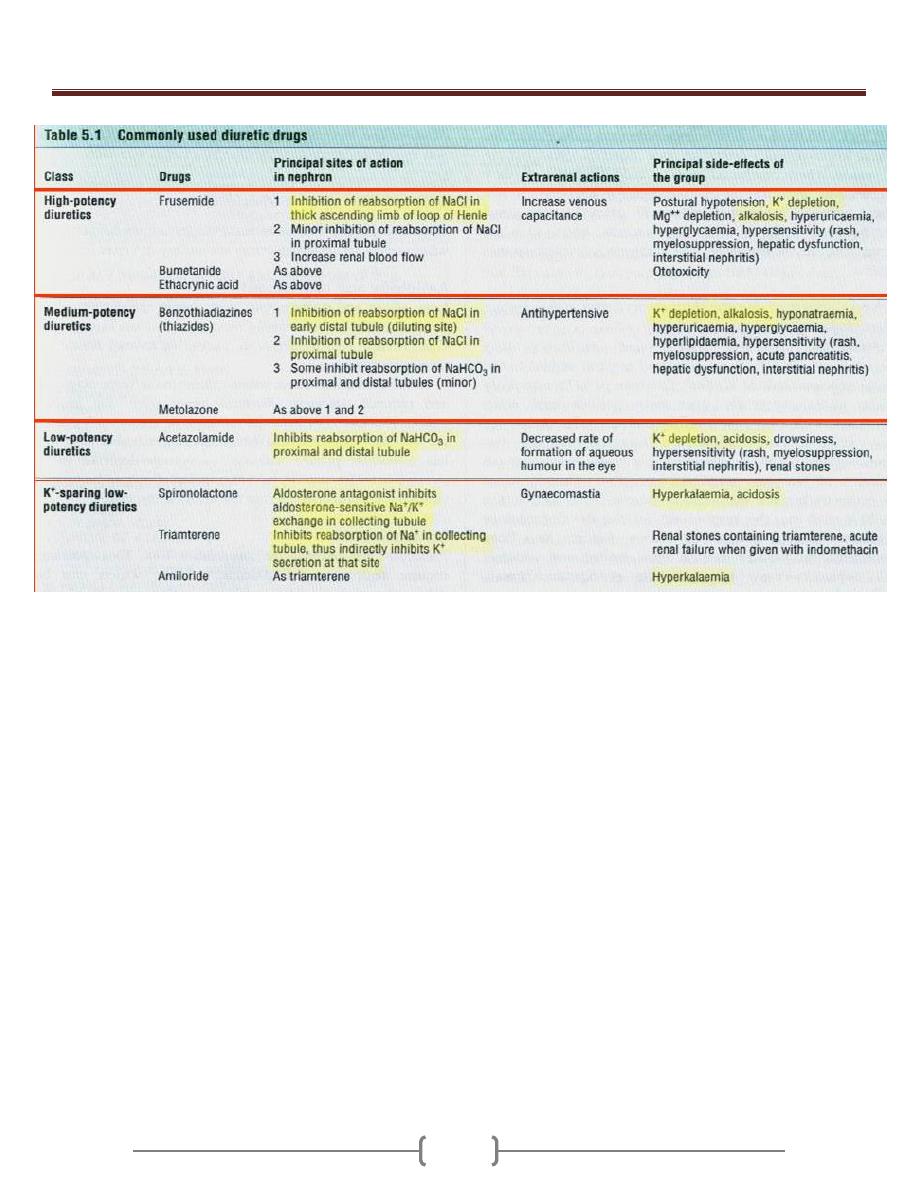
Unit 7 - Clinical biochemistry and metabolism
92