
1
Fifth stage
Medicine
Lec-7
د. بشار
8/12/2015
DEMYELINATING DISEASES
MULTIPLE SCLEROSIS
In multiple sclerosis, one of the most common neurological causes of long-term disability,
the myelin-producing oligodendrocytes of the central nervous system are the target of
recurrent cell-mediated autoimmune attack.
In the UK the prevalence is 120 per 100 000 of the population, with an annual incidence of
around 7 per 100 000. The lifetime risk of developing multiple sclerosis is about 1 in 400.
The incidence is higher in temperate climates and in Northern Europeans, and the disease is
about twice as common in women as men.
Aetiology
Epidemiological and genetic evidence suggests that multiple sclerosis is caused by an
interplay of multiple genetic and environmental factors. The incidence varies with latitude,
being low in equatorial areas and higher in the temperate zones of both hemispheres, with
people retaining the risk of the zone in which they grew up, indicating that environmental
exposure during growth and development are impotant.
The prevalence has also been found to correlate with various environmental factors, such
as sunlight exposure, vitamin D and exposure to EBV,although it is unclear exactly how all of
these factors interact to cause the disease.
An immune mechanism is suggested by increased levels of activated T lymphocytes in the
CSF, and increased immunoglobulin synthesis within the central nervous system .
The risk of familial recurrence is 15%, with highest being for first-degree relatives (age-
adjusted risk: 4 -5 %for siblings2-3% for parents or offspring) and a monozygotic twin
concordance of 30%. The inheritace appears to be polygenic, with influences from the HLA
regional, IL-7 R (interleukine -7 receptor ), IL – 2 R (interleukine – 2 receptor),CLEC16A( C-
type lectin domain family 16 member A ).
Pathology
An attack of central nervous system inflammation in multiple sclerosis starts with the entry
of activated T lymphocytes through the blood-brain barrier. These recognise myelin-derived
antigens on the surface of the nervous system's antigen-presenting cells, the microglia, and
undergo clonal proliferation. The resulting inflammatory cascade releases cytokines and
initiates destruction of the oligodendrocyte-myelin unit by macrophages.

2
Histologically, the characteristic lesion is a plaque of inflammatory demyelination
occurring most commonly in the periventricular regions of the brain, the optic nerves and
the subpial regions of the spinal cord. Initially, this is a circumscribed area of disintegration
of the myelin sheath, accompanied by infiltration by activated lymphocytes and
macrophages, often with conspicuous perivascular inflammation. After an acute attack,
gliosis follows, leaving a shrunken grey scar .
Pathophysiology
Much of the initial acute clinical deficit is caused by the effect of inflammatory cytokines
upon transmission of the nervous impulse rather than structural disruption of the myelin,
which explains the rapid recovery of some deficits and probably the efficacy of
corticosteroids in ameliorating the acute deficit.
However, the myelin loss that results from an attack reduces the efficiency of impulse
propagation or causes complete conduction block, which impairs the efficiency of central
nervous system functions. Inflammatory mediators released during the acute attack
(particularly nitrous oxide) probably also initiate axonal damage, which is a feature of the
latter stages of the disease.
In established multiple sclerosis there is progressive axonal loss, probably due to direct
damage to axonal integrity by the inflammatory mediators released in acute attacks and
subsequently the loss of neurotrophic factors from oligodendrocytes. This axonal loss is the
cause of the phase of the disease in which there is progressive and persistent disability .
Clinical features
Demyelinating lesions cause symptoms and signs that usually come on subacutely over days
or weeks and resolve over weeks or months, although rarely a stroke-like presentation may
occur.
After a variable interval there may be a recurrence, often within 2 years. Frequent relapses
with incomplete recovery indicate a poor prognosis, and in many patients a phase of
secondary progression, caused by secondary axonal degeneration, supersedes the phase of
relapse and remission.
In a minority of patients, there may be an interval of years or even decades between
attacks, and in some, particularly if optic neuritis is the initial manifestation, there is no
recurrence. Some presentations, such as optic neuritis with purely sensory relapses, have a
good prognosis .
3
The physical signs observed in multiple sclerosis depend on the anatomical site of
demyelination. Combinations of spinal cord and brain-stem signs are common, may be
with evidence of previous optic neuritis in the form of an afferent pupillary deficit.
Significant intellectual impairment is unusual until late in the disease, when loss of frontal
functions and impairment of memory are common
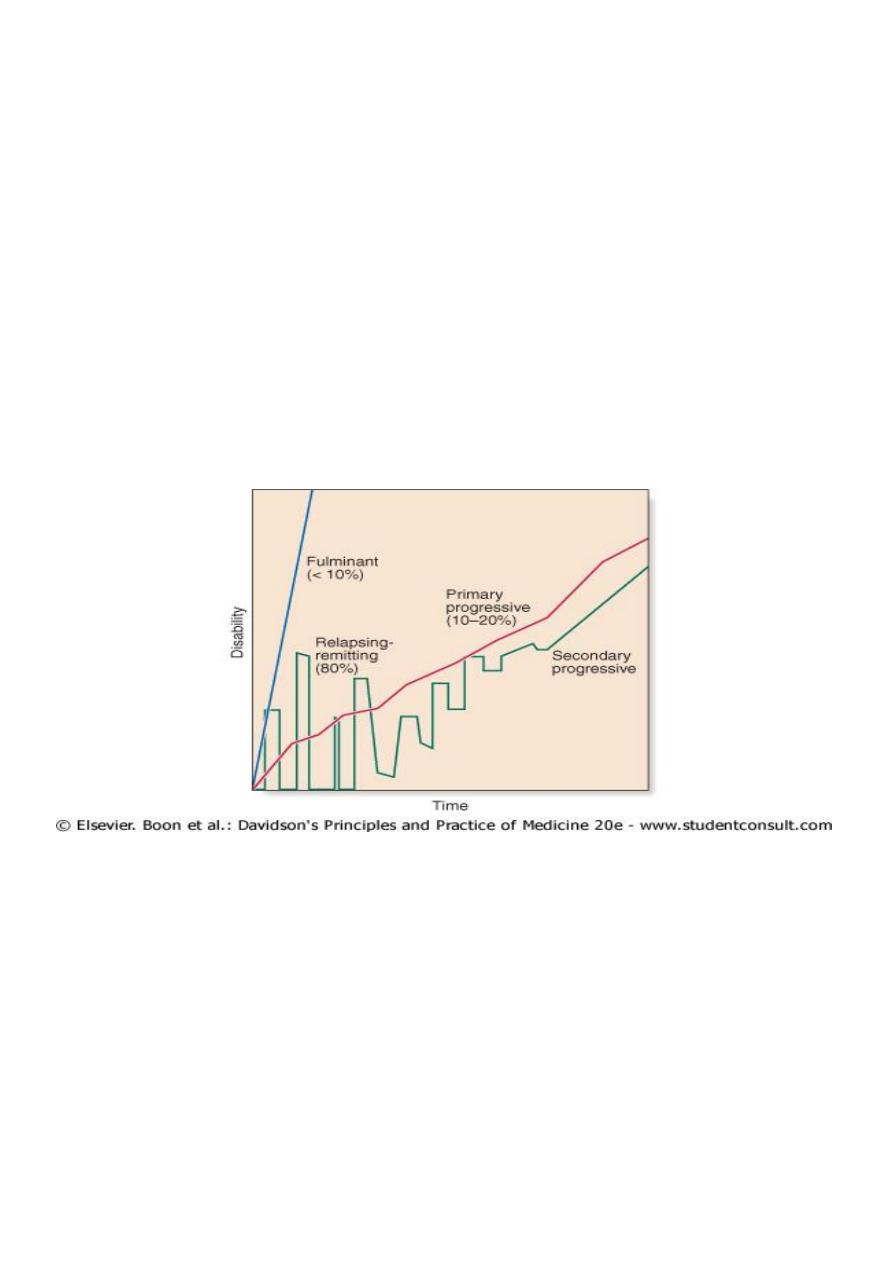
Course
Around 80% of patients have a relapsing and remitting clinical course of episodic
dysfunction of the central nervous system with variable recovery. Of the remaining 20%,
most follow a slowly progressive clinical course, with a tiny minority who have a fulminant
variety leading to early death.
The peak age of onset is in the fourth decade, with onset before puberty or after the age of
60 years being rare.
Common presentations of multiple sclerosis
Optic neuritis
Relapsing and remitting sensory symptoms
Subacute painless spinal cord lesion
Acute brain-stem syndrome
Subacute loss of function of upper limb (dorsal column deficit )
6th cranial nerve palsy

4
Optic neuritis
In about 25 percent of all MS patients (and in a larger proportion of children), the initial
manifestation is an episode of optic neuritis. Characteristically, over a period of several
days, there is partial or total loss of vision in one eye. Many patients, for a day or two
before the visual loss, experience pain within the orbit, worsened by eye movement or
palpation of the globe.
Usually a scotoma involving the macular area and blind spot (cecocentral) can be
demonstrated, but a wide variety of other field defects may occur, rarely even hemianopic
involvement sometimes homonymous).
In some patients, both optic nerves are involved, either simultaneously or, more
commonly, within a few days or weeks of one another .
Papillitis vs Retrobulbar neuritis
About half of patients with optic neuritis recover completely, and most of the remaining
ones improve significantly, even those who present initially with profound visual loss and,
later, pallor of the optic disc.
One-half or more of adult patients who present with optic neuritis will eventually develop
other signs of MS.
Relapsing and remitting sensory symptoms
Transient facial hypesthesia or anesthesia
Dull aching pain in the low back
Sharp, burning, poorly localized, or lancinating-radicular pain, localized to a limb or
discrete part of the trunk
Paresthesias or numbness of an entire arm or leg
Spinal cord lesion ( Transverse Myelitis )
Symmetrical or asymmetrical paraparesis or paraplegia, ascending paresthesias, loss of
deep sensibility in the feet, a sensory level on the trunk, sphincteric dysfunction, and
bilateral Babinski signs.
Acute brain-stem syndrome
Diplopia
Myokymia or paralysis of facial muscles,
Deafness, tinnitus,vertigo
Cerebellar signs
5
Test for visual field defects (confrontation test) :
Temporal disc atrophy :
Sensory disturbances :
Other symptoms and syndromes suggestive of CNS demyelination :
Afferent pupillary defect and optic atrophy (previous optic neuritis
)
Lhermitte's symptom (tingling in spine or limbs on neck flexion
)
Progressive non-compressive paraparesis
Partial Brown-Séquard syndrome
Internuclear ophthalmoplegia with ataxia
Holmes (rubral ) tremor ( RT + PosT + IT, mainly proximal, disabling.
Trigeminal neuralgia (under the age of 50 )
Recurrent facial palsy

6
INO :
Paroxysmal symptoms (trigeminal neuralgia) :
Ataxia &Incoordination :
Motor disturbances
)
central paresis, spasticity,
abnormal fatigability)
7
Central paresis (right hyperreflexia) :
Autonomic dysfunction
:
urinary/fecal incontinence, sexual
dysfunction :
Behavioral changes :
Investigations
There is no specific test for multiple sclerosis, and the results of investigation are taken in
conjunction with the clinical picture in making a diagnosis of varying probability.
The clinical diagnosis of multiple sclerosis should be supported by investigations to exclude
other conditions, provide evidence for an inflammatory disorder and identify multiple sites
of neurological involvement
CSF
The cerebrospinal fluid (CSF) is commonly abnormal, with mild lymphocytosis or a slightly
increased protein concentration, especially if examined soon after an acute relapse.

8
CSF protein electrophoresis shows the presence of discrete bands in the immunoglobulin G
(IgG) region ( oligoclonal bands) in 70 - 90% of patients between attacks. The antigens
responsible for these antibodies are not known.
Oligoclonal bands are not specicfic to MS but denote intra thecal inflammation.
Evoked Potentials EP :
If clinical evidence of a lesion exists at only one site in the central nervous system, a
diagnosis of multiple sclerosis cannot properly be made unless other regions have been
affected subclinically, as detected by the electrocerebral responses evoked by one or more
of the following:
monocular visual stimulation with a checkerboard pattern (visual evoked potentials -
VEP);
monaural stimulation with repetitive clicks ( brainstem auditory evoked potentials -
BAEP);
electrical stimulation of a peripheral nerve (somatosensory evoked potentials - SSEP).
VEP measurement : VEP :
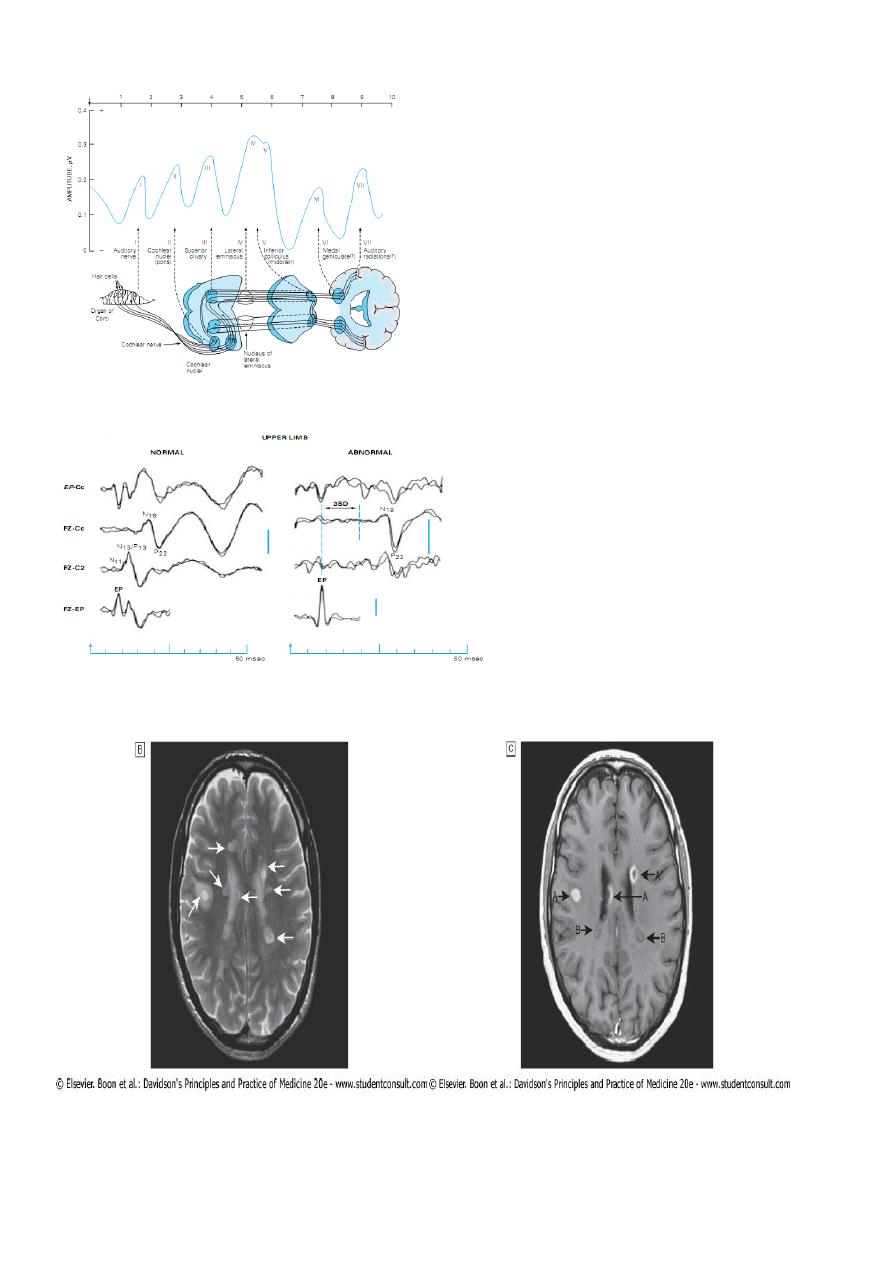
9
BAEP :
SSEP :
MRI may also detect subclinical lesions and has become nearly indispensible in confirming
the diagnosis

11
Other investigations :
CXR
Serum angiotensin converting enzyme( ACE) Sarcoidosis
Serum B12
Antinuclear Antibodies – SLE
Antiphospholipid Antibodies
THE MACDONALD CRITERIA FOR THE DIAGNOSIS OF MULTIPLE SCLEROSIS

11
Management :
This involves treatment of the acute episode,prevention of future relapses,treatment of
complications and management of the patient disability.
The acute episode :
In a function threatening exacerbation of MS pulses of high dose methylprednisolone ,
either I.V. (1 gm daily for 3 days or orally (500 mg daily for 5 days),shorten the duration of
the episode.
Pulsed steroids also have some effect in reducing spasticity.
Prolonged administration of steroids does not alter the long-term outcome and is therefore
avoided.
Pulses of steroids can be given up to three times in a year but their administration should
be restricted to those with significant function-threatening deficits.
Prophylaxis to prevent the occurrence of steroid –induced osteoporosis should be
considered.
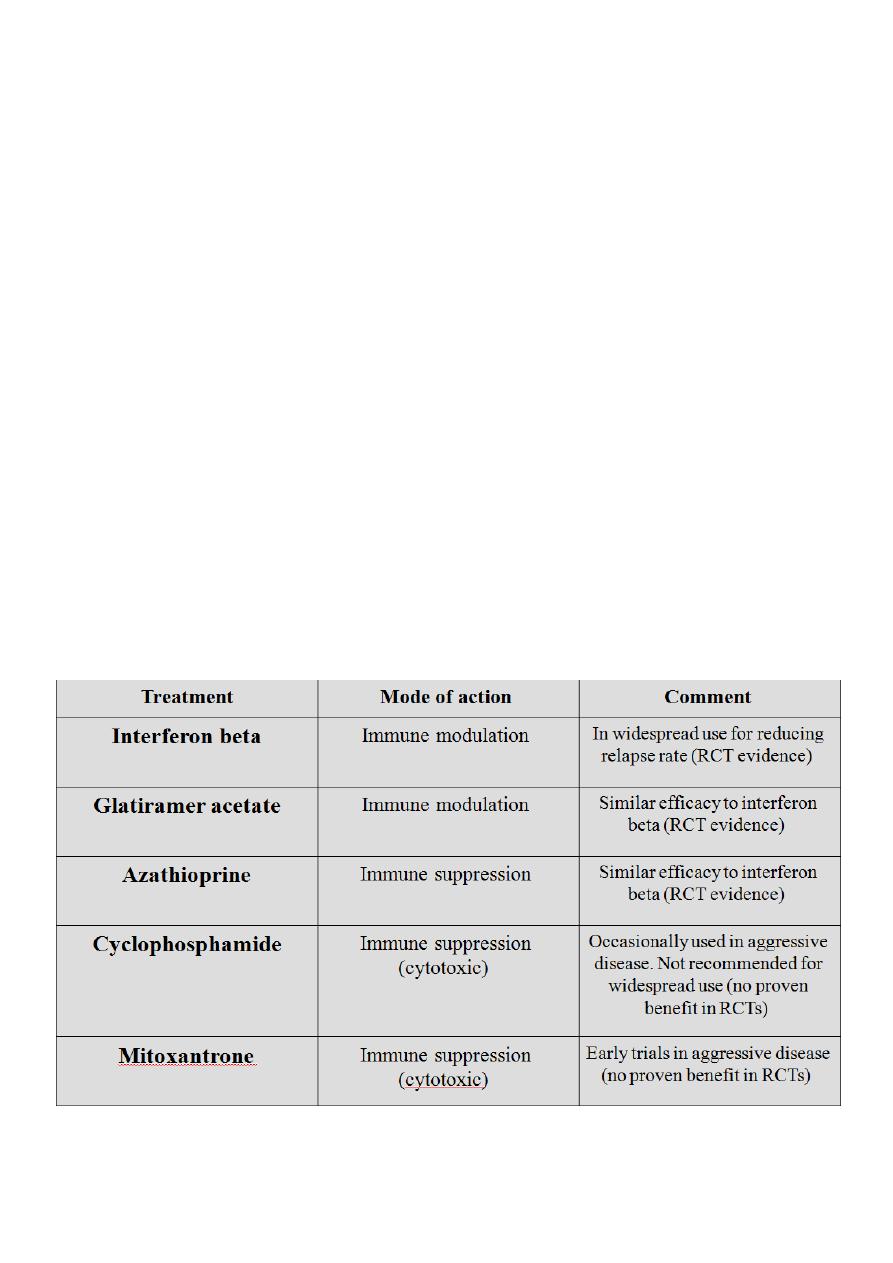
Preventing relapses :
Immunosuppressive agents including azathioprine appear to reduce the risk of relapses
and improving long-term outcome.
In relapsing and remitting multiple sclerosis, subcutaneous or intramuscular interferon
beta-1a/b reduces the number of relapses by some 30%, with a small effect on long-term
disability ; glatiramer acetate has similar effects. Glatiramer is a polymer of 4 aminoacids
found inMBP which is thought possibly to act as a decoy for the immune reponse in
patients with MS.
The most common side effects of interferons are a flu-like syndrome and (in the case of
interferon b-1b) injection site reactions. Glatiramer acetate is generally tolerated well, but
it may produce erythema at the sites of injection, and about 15% of patients experience
transient episodes of flushing, dyspnea, chest tightness,
palpitations, and anxiety after injections.
All three of these agents are approved for use in relapsing-remitting multiple sclerosis and
are available by prescription. They are expensive, but their cost must be balanced against
the reduced need for medical care and reduced time lost from work that follows their use.
The recently introduced agent natalizumab is probably somewhat more effective than both
but is usually preserved for patients with more aggressive disease, along with the less
proven therapies as mitoxantrone and cyclophosphamide.
12
Special diets including gluten-free, linoleic acid supplements or hyperbaric oxygen therapy
are popular with patients, but are of no proven benefit.
Management
Appropriate treatment of primary or secondary progressive multiple sclerosis is less well
established.
Recent studies suggest that interferon b-1b (and probably
interferon b-1a) are effective in reducing the progression rate as determined clinically and
by MRI in secondary progressive disease, but there is only limited experience with
glatiramer acetate in this setting.
Treatment with cyclophosphamide, azathioprine, methotrexate, cladribine, or
mitoxandrone may help to arrest the course of secondary progressive disease, but studies
are inconclusive.
Pulse therapy with high-dose intravenous methylprednisolone (1 g/d once a month) is also
sometimes effective and may carry a lower risk of long-term complications than the
cytotoxic drugs.
DISEASE-MODIFYING TREATMENTS IN MS :
13
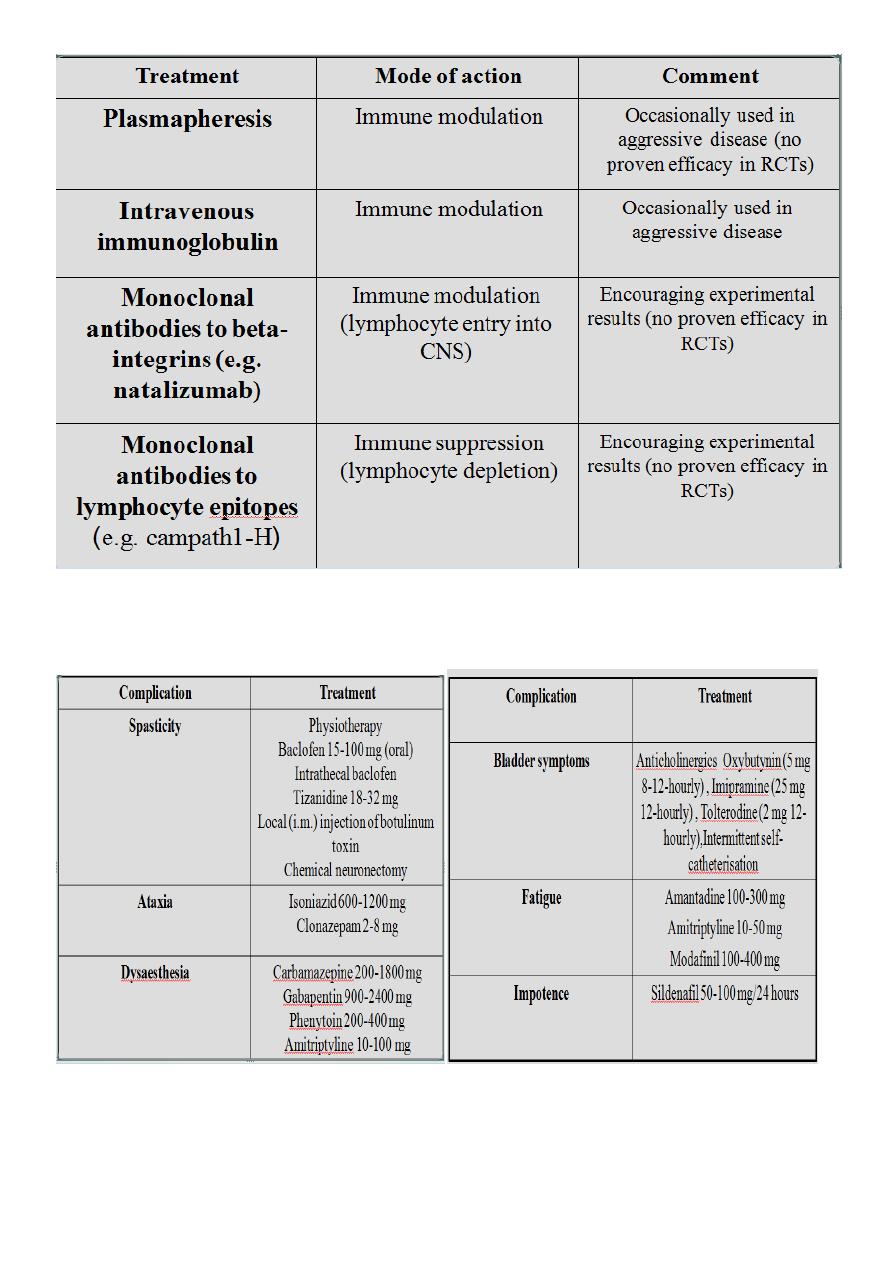
TREATMENT OF COMPLICATIONS OF MULTIPLE SCLEROSIS :

14
Prognosis :
The outlook is difficult to predict with confidence in any individual patient, especially early
in the disease.
Furthermore, the ability to diagnose disease at an earlier stage means that older studies
may not reliably reflect the outcome of those diagnosed with modern techniques.
About 15% of those having one attack of demyelination do not suffer any more events,
whilst those with relapsing and remitting multiple sclerosis have, on average, 1-2 relapses
every 2 years.
Approximately 5% of patients die within 5 years of onset, whilst others have a very benign
outcome. Overall, after 10 years about one-third of patients are disabled to the point of
needing help with walking, rising to about 50% after 15 years.
Features that tend to imply a more favorable prognosis include :
Female sex,
Onset before age 40 .
Presentation with visual or somatosensory, rather than pyramidal or cerebellar
dysfunction.
ACUTE DISSEMINATED ENCEPHALOMYELITIS
This is an acute, usually monophasic, demyelinating condition in which there are areas of
perivenous demyelination widely disseminated throughout the brain and spinal cord. The
illness may apparently occur spontaneously but often occurs a week or so after a viral
infection, especially measles and chickenpox, or following vaccination, suggesting that it is
immunologically mediated.
Clinical features :
Headache, vomiting, pyrexia, confusion and meningism may be presenting features, often
with focal or multifocal brain and spinal cord signs. Seizures or coma may occur. A minority
of patients who recover have further episodes .
Investigations :
MRI shows multiple high-signal areas in a pattern similar to that of multiple sclerosis,
although often with larger areas of abnormality. The CSF may be normal or show an
increase in protein and lymphocytes (occasionally over 100 × 106 cells/l).
Oligoclonal bands may be found in the acute episode but do not persist upon recovery,
unlike in multiple sclerosis. The differential diagnosis from a first severe attack of multiple
sclerosis may be difficult.

15
Management :
The disease may be fatal in the acute stages but is otherwise self-limiting. Treatment with
high-dose intravenous methylprednisolone, using the same regimen as for a relapse of
multiple sclerosis, is recommended.
Neuromyelitis optica :
The concurrence of transverse myelitis and bilateral optic neuritis in some patients has
been recognized in some patients,and these clinical manifestations are more common in
MS which occurs in Asia.
The majority of these cases are associated with an antibody to water channel,aquaporin 4,
which is found in cells near the ventricular system of the brain.
Patients typically have brain MRI scans which are either normal or have high signal lesions
restricted to the region of the ventricular system.Spinal MRI scans show lesions which are
typically longer than 3 spinal segments(unlike the lesions of ms).Clinical deficits tend to
recover less well than those of MS,and the disease may be more aggressive with more
frequent relapses.Teatment with immunosuppressive agents such as steroids,azathioprine
or cyclophosphamide and or plasmapheresis seems to be more effective than in MS.
ACUTE TRANSVERSE MYELITIS :
Transverse myelitis is an acute, often monophasic, inflammatory demyelinating disorder
affecting the spinal cord over a variable number of segments. Patients may be of any age
and present with a subacute paraparesis with a sensory level, often with severe pain in the
neck or back at the onset. MRI is needed to distinguish this from a compressive lesion of
the spinal cord. CSF examination shows cellular pleocytosis, often with polymorphs at the
onset, and oligoclonal bands are usually absent. Treatment is with high-dose intravenous
methylprednisolone. The outcome is variable; in some cases, near-complete recovery
occurs despite a severe initial deficit. Some patients who present with acute transverse
myelitis go on to develop multiple sclerosis in later years.