Medicine
HAEMATOLOGY
.-
Total Haematology Lec : 12
Al-Madena Copy
Lecture #1


1
Hemolytic anemias
Definition:Anemias that result from shortening of RBC life span,RBC
destruction could be extravascular or intravascular
Etiology
congenital
Acquired
1- Membrane abnormalities
• Hereditary spherocytosis
• Hereditary elliptocytosis
1- Immune
•
Autoimmune Warm antibody
Cold antibody
•Alloimmune
2-Haemoglobinopathies
• Lack of haemoglobin chain
synthesis Thalassaemias
• Amino acid substitution on the
haemoglobin chain Haemoglobin S.
C, D
2- Non-immune
•
Mechanical
- Artificial cardiac valves
- Burns
- Microangiopattlic
- March haemoglobinuria
•
Infections
Clostridium perfringens, malaria
•
Drugs, chemicals
3- Red cell enzyme detects
• Glucose-B-phosphate
dehydrogenase deficiency
3- Paroxysmal nocturnal
haemoglobinuria (PNH).

2
Approach To Hemolytic anemia
A- Prove The Presence of Hemolysis (Evidence of hemolysis):
q
Clinical Features: anemia +jaundice.
q
Laboratory Tests
Low Hb
Increased reticulocyte count and percentage.
Indirect hyperbilirubinemia
raised s.LDH,
increased urinary urobilinogen.
Low serum haptoglobin.
Hemosiderinurea, hemoglobinurea in cases of intravascular hemolysis.
B-Find The Cause Of hemolysis
1- Blood Film Morphology: Target Cells,Sickle Cells,Heinz Bodies, Blister
Cells, Fragmented RBC, Spherocytes.
2-Hemoglobin Electrophoresis: for
Hemoglobinopathies.
3- Osmotic Fragility test for Spherocytosis
4- Enzyme assay for Enzymopathies.
5- Coomb
,
s test for immune hemolysis.
6- Ham
,
s test for PNH.
25 years old female accidentally discover she had splenomegally while she
had biliary colicy pain ,patient has no symptomes apart her new biliary colic no
history of transfusion, and she has family history of early cholecystectomy

3
CBC: showed spherocytosis, increase corrected retic 5% ,T.S.B 2mgldl,coombs
was negative
Hereditary spherocytosis
This is an autosornal dominant disorder in which the principal
abnormality appears in red cell membrane protein.
Approximately 25% of patients have no family history.
The erythrocyte envelope is abnormally permeable and the sodium pumps
are overworked.
ü
The exact nature of the red blood cell defect may vary from family to
family.
The severity of the disorder is very variable even within an affected
family.
The erythrocytes lose their biconcave shape, become spherical and are
more susceptible to osmotic lysis.

4
These spherocytes are destroyed almost exclusively by the spleen.
Haemolysis is mainly extravascular.
Diagnosis &Treatment
Clinical Features:
q
The severity of anemia is variable from asymptomatic to transfusion
dependent anemia.Jaundice is also variable,as well as splenomegaly.
q
Complications include
1- Crises (hemolytic, megaloblastic, and aplastic).
2- Pigment Gall stones.
Lab Tests:
q
Evidence of Hemolysis: Anemia, Reticulocytosis, raised S.LDH…
q
Spherocytes on blood film.
q
+ve Osmotic Fragility Test.
q
-ve Coombs Test.
Treatment :
1-Blood Transfusion.
2-Folic acid.
3-Splenectomy for moderate to severe cases.
20 years old male presented with abdominal pain followed by dark color
urine .he had history of flu-like illness with history of ingestion of
trimethprim.
family noticed that he
become pale within few hours ago.
CBP:Hb 7 gm/l ,retic 15%,with presence of Heinz body,and blister cell,and
spherocytes

5
Glucose 6 Phosphate Dehydrogenase Deficiency
q
G6PD Enzyme is the first one in the hexosmonophosphate shunt, the
function of this shunt is to service the enzymes glutathione reductase
and glutathione peroxidase, which protect the red cells against damage
due to oxidation.
G6PD deficiency
q
The deficiency is inherited as an X-linked disorder
q
Oxidant damage of RBC followed by intavascular hemolysis is induced
by:
1- Infections .
2- Ingestion of Fava Beans.
3-Oxidant drugs like: sulfa, dapsone, antimalarial, chloramphenicol…..,
4- Surgery.
Clinical manifestations:
1- Most cases present
with fever, rapid anemia, jaundice
and deep colored urine.
2- Rarely the hemolysis is chronic.
3- Neonatal jaundice.
G6PD deficiency
Lab Tests:
1- Anemia, reticulocytosis,
indirect hyperbilirubinemia…
2- Blister RBC on blood film,
and Heinz bodies.

6
3- Hemoglobinurea and later
hemosideriurea.
4- Enzyme assay is useful
after recovery.
Treatment:
1- Avoid fava beans and oxidant drugs.
2- For the hemolytic episode: stop the offending factor,
blood transfusion, folic acid.
HAEMOGLOBINOPATHIES
q
The haemoglobinopathies can be classified into two subgroups:
1- Where there is an alteration in the amino acid structure of the polypeptide
chains of the glohin fraction of haemoglohin, commonly called the abnormal
haemoglohins: the best-known example is haemoglobin S found in sickle-cell
anaemia.
2- Where the polypeptide chain production is impaired or absent for a
variety of reasons: these are the Thalassemias.
Hemoglobin Structure and Production
q
Fetal hemoglobin, HbF (α2γ2) switches to adult forms HbA (α2ß2) and
HbA2 (α2δ2) at 3-6 months of life.
q
HbA constitutes 97% of adult hemoglobin.
q
HbA2 constitutes 3% of adult hemoglobin.
q
4 α genes are located on chromosome 16.

7
q
2 ß genes are located on chromosome 11.
q
There is the possibility of mixed defects
e.g. ß-thalassemia minor and sickle cell (HbS) trait.
15years old child presented with ascitis and leg oedemea .he referred by
surgeon planning for splenectomy due to huge spleen.
Pateint transfusion dependent since childhood and his brother dead on his
second decade.
On examination: anemic, with slate grey skin and puffy face, leg
oedema,Ascitis,hepatosplenomegally
CBC:Hb:7.8,retic 7%,NRBC10/50cell,MCV 56,hypochromic microcytic
WBC :3.5x109
Platelet 40x109
Pregnant lady with 24wks gestation referred by her gynecologist as history of
increasing anemia and and needs of transfusion. Patient denied any
previous history of transfusion and her hemoglobin level since childhood
around 10gm/l with family history of anemia
ex.patient pale ,not jaundice ,no organomegaly
CBC:Hb 8gm/l,retic 5%,MCV 60
Hypochromic microcytic
S.Ferritin was normal

8
THALASSEMIAS
HETEROZYGOU ß THALASSEMIA
: ß-Thalassemia Minor
q
Common condition in Mediterranean Basin ,Africa,Asia
Clinical Presentation
1. Mild or no anemia.
2. Spleen sometimes is palpable.
3. May be masked by Fe deficiency and sometimes confused with iron
deficiency anemia.
Diagnosis
1- Hb 9-12 g/dL, MCV < 70
2- Microcytosis +/– hypochromia,target cells present,basophilic stippling
usually present.
3- Hb electrophoresis: Hb A2 increased to 3.5-5% (normal 1.5 - 3.5%), 50%
of individuals have slight increase in HbF.
Management
1. Add folic acid.
2. Patient and family should receive genetic counseling.
HOMOZYGOUS ß THALASSEMIA (ß-Thalassemia Major)
Pathophysiology
Ineffective beta chain synthesis due to point mutation in the beta gene
promoter or enhancer on chromosome 11, excess alpha chains relative to

9
beta chain leading to ineffective erythropoiesis and hemolysis of RBC,
compensatory increase in HbF
Clinical Presentation
q
Start presenting at 3-6 months because of replacement of HbF by HbA
q
Severe anemia developing in the first year of life
q
Jaundice
q
Stunted growth and development (hypogonadal dwarf)
q
Gross hepatosplenomegaly (extramedullary hematopoiesis)
q
Skeletal changes (expanded marrow cavity)
• Skull x-ray has “hair-on-end” appearance
• Pathological fractures common
q
Evidence of increased Hb catabolism (e.g. gallstones)
q
Death from
• Untreated anemia .
• Infection (early).
• Iron overload (late, secondary to transfusions), usually20-30
years old.
PATHOPHYSIOLOGY OF B-THALASSEMIA MAJOR
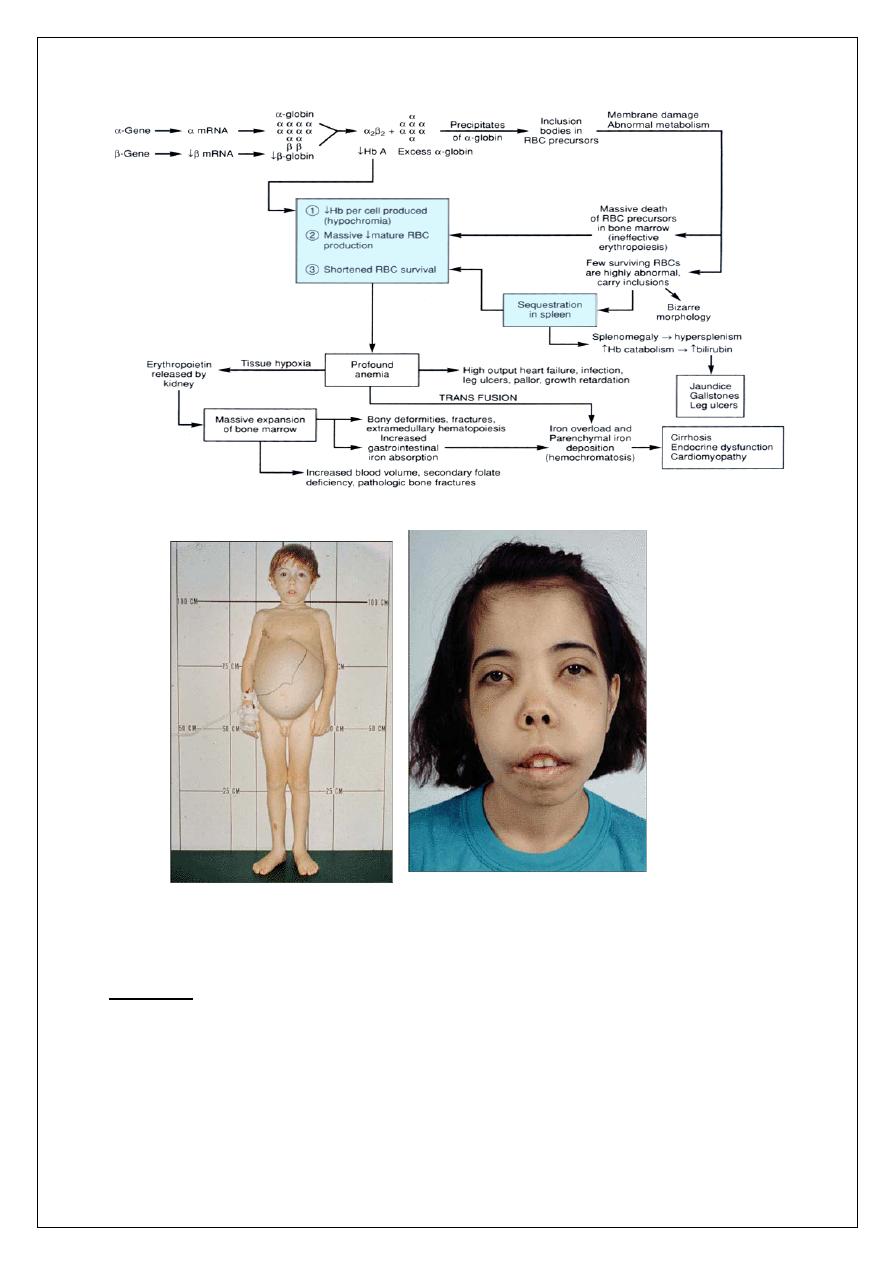
10
ß-Thalassemia Major
Diagnosis
1- Hemoglobin 4-6 g/dL.
2- Peripheral blood: hypochromic microcytosic, increased reticulocytes,
basophilic stippling, target cells.

11
- Postsplenectomy blood film shows Howell Jolly bodies, Nuleated RBC,
and thrombocytosis.
3- Hb electrophoresis
• Hb A: 0-10% ( normal > 95%)
• Hb F: 90-100%
4-DNA analysis.
Management (ß-Thalassemia Major)
1- Blood transfusions to ensure growth and decrease skeletal deformities ,try
to keep Hb > 10 gm/dL, add folic acid.
2- Fe chelators to prevent iron overload like desferrioxamine,
deferiprone.defrasirox.
3- Ensure good nutrition and try to minimize the frequency of infectious
episodes. Infections should be treated adequately.
4- Allogeneic Bone marrow transplant (if suitable donor).
5- Splenectomy for mechanical problems and hypersplenism.
6- Genetic counseling for prevention

12
ALPHA THALASSEMIAS
Pathophysiology
q
Autosomal recessive
q
Deficit of alpha chains
q
4 grades of severity depending on the number of defective alpha genes
1 - silent: αα/ α-
2 - trait: αα/-- or α-/ α-
3 - Hb H Disease (presents in adults) : α-/--
4 - Hb Bart’s (hydrops fetalis): --/--
q
Hb Bart’s made of 4 gamma chains; not compatible with life
q
Hb H excess of beta chains, is unstable, and leads to inclusion bodies
Diagnosis
1- Peripheral blood film: microcytes, hypochromia, occasional
target cells, screen for Hb H inclusion bodies.
2- Hb electrophoresis not diagnostic.
3- DNA analysis using alpha gene probe.
Management: same as beta thalassemia.
PATHOPHYSIOLOGY OF ALPHA THALASSEMIAS

13
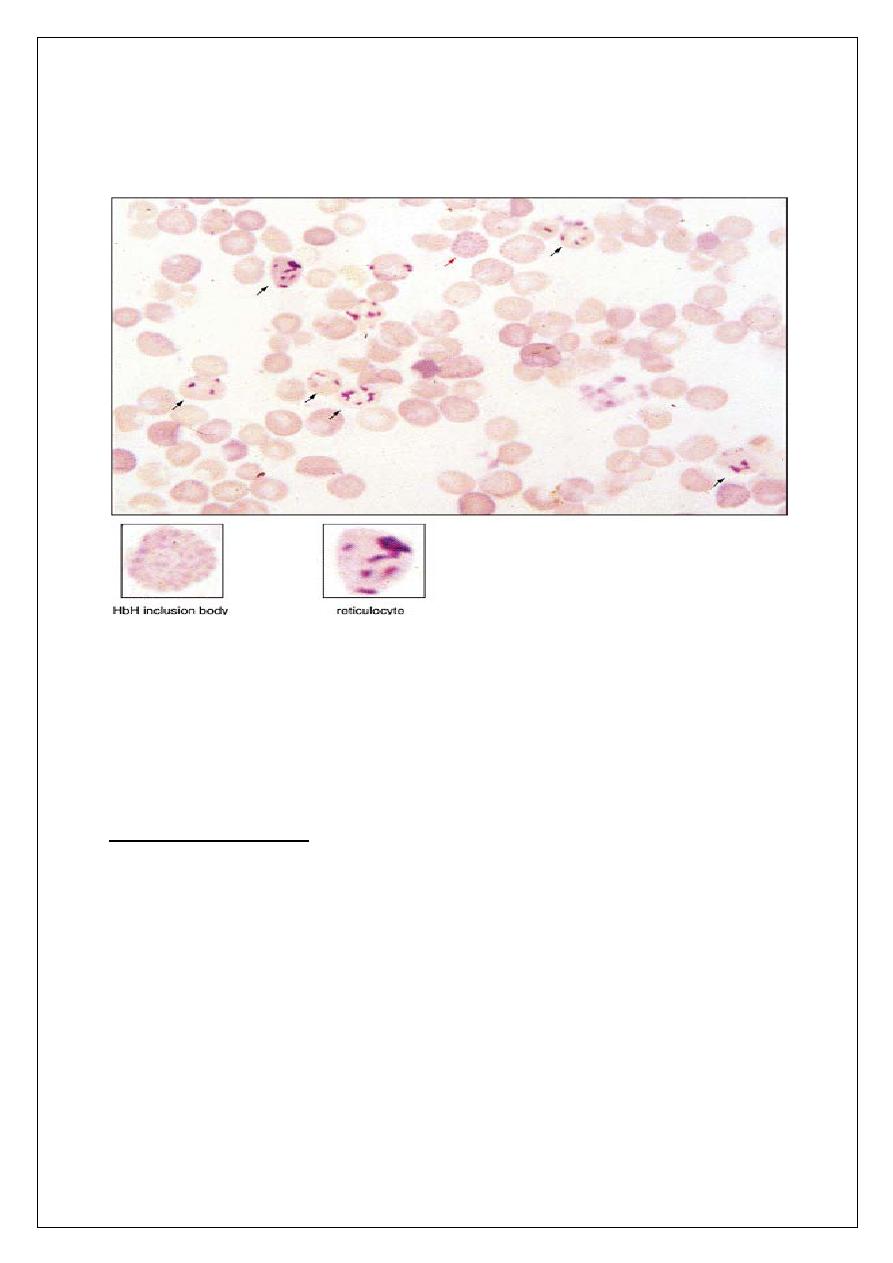
14
HEMOGLOBIN H INCLUSION BODIES
Sickle Cell anemia
q
Autosomal recessive
q
Amino acid substitution of valine for glutamate in position 6 of beta
globin chain.

15
*It has a wide geographical distribution.
Sickle Cell anemia
Mechanisms of Sickling
q
At low PO2, deoxy Hb S polymerizes, leading to rigid crystal-like rods
that distort membrane = SICKLES
q
The PO2 level at which sickling occurs is related to the % of Hb S
present
- If the patient is heterozygous (Hb AS), the sickling phenomenon occurs
at a PO2 of 40 mmHg
- If the patient is homozygous (Hb SS), sickling occurs at 80 mmHg
q
Sickling is also aggravated by
1. Acidosis.
2. Increased CO2.
3. Increased 2,3-DPG.
4. Increased temperature and osmolality.
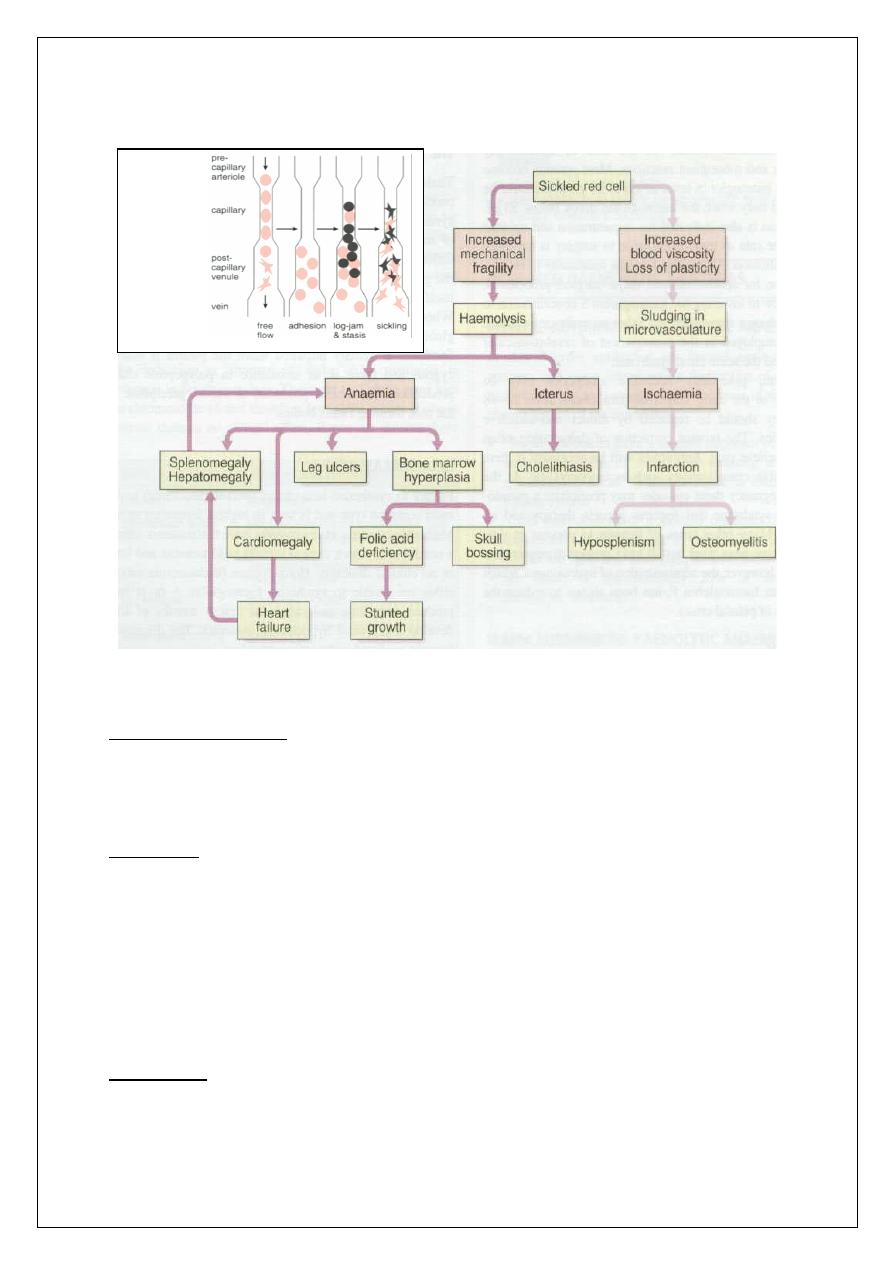
16
Clinical Consequences Of SCA
Heterozygous SCA: Hb S Trait
Clinical presentation:
• the patient will appear normal except at times of extreme hypoxia and
infection, elderly patients may suffer from loss of renal concentration ability.
Diagnosis:
1- Hb level is normal
2- Peripheral blood: normal except for a few target cells
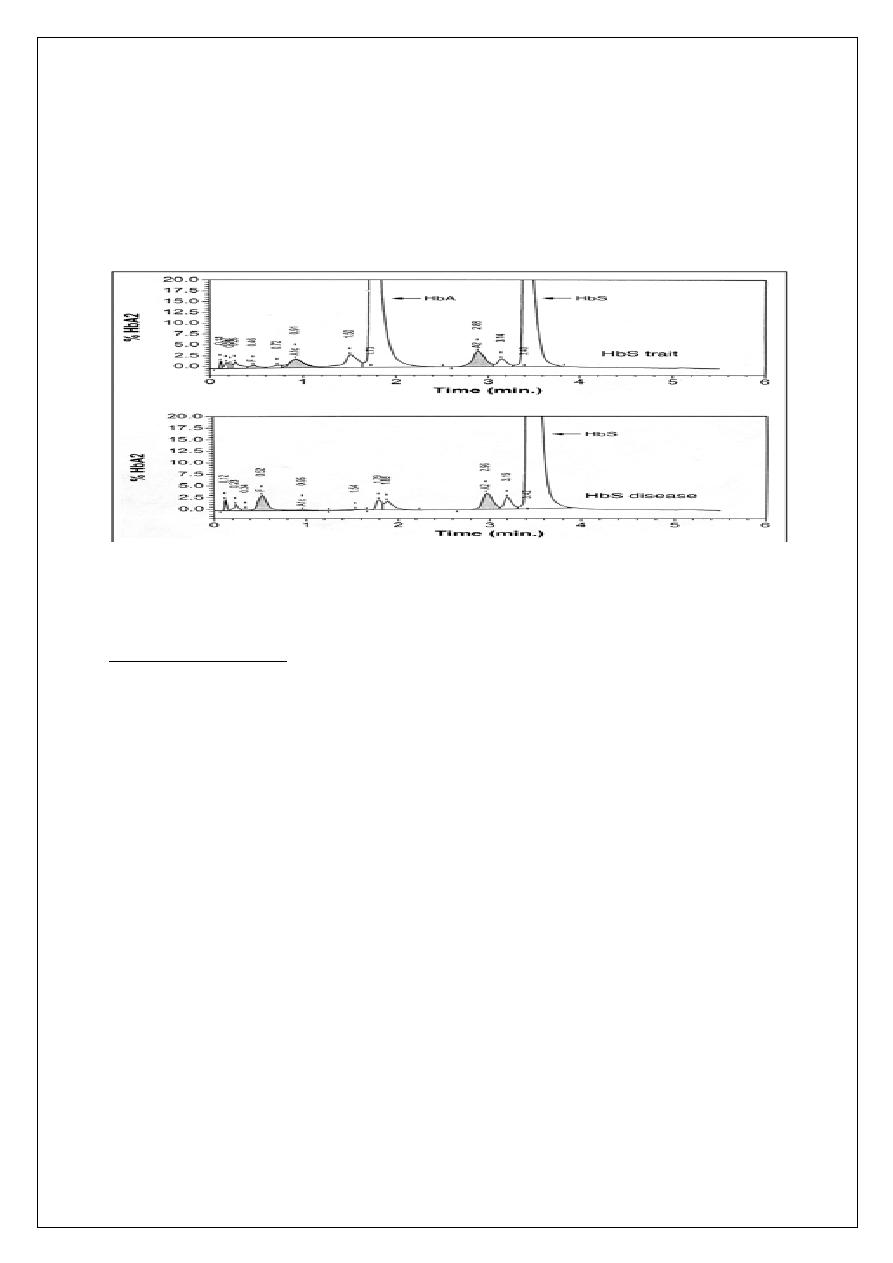
3- Hb electrophoresis (confirmatory test): Hb A fraction
of 65% ; Hb S fraction of 35%
Treatment:
1- Avoid hypoxia during flying and surgery.
17
2- Folic acid for pregnant.
3- Genetic counseling.
HEMOGLOBIN ELECTROPHORESIS IN SCA
Homozygous SCA: Hb SS Disease
Clinical presentation
1- Chronic hemolytic anemia with jaundice in the first year of life.
2- Retarded growth and development +/– skeletal changes.
3- Susceptibility to infections by encapsulated organisms due to
hyposplenism.
4- Spleen enlarged in children and atrophic in adults.
5- Crises:
Vaso - occlusive crises (infarction) leading to pain, fever , leukocytosis,
acidosis& dehydration. Any organ or tissue can be involved.
Hyperhemolytic crises associated malaria.
Sequestration crises presenting with anemia and rapidly enlarging spleen or
liver.

18
Aplastic crisis due to parvovirus infection or folate deficiency, leading to
rapid anemia and reticulocytopenia.
Acute Chest Syndrome presenting as fever, chest pain, cough and hypoxia.
6- Iron overload due to repeated blood transfusion (less likely compared to
Thalassemia).
7-Gall stones,leg ulcers.
Homozygous SCA: Hb SS Disease
Diagnosis
1- Peripheral blood: sickled cells,target cells, reticulocytosis.
2- Indirect hyperbilirubinemia, raised s.LDH
3- Screening test: sickle cell preparation searching for sickling phenomenon.
4- Hb electrophoresis (confirmatory test): Hb S fraction > 80%,the rest is Hb F.
Management (Homozygous SCA: Hb SS Disease)
1- Prevention Of Sickling Attacks:
• Avoid conditions that favor sickling (hypoxia, acidosis, dehydration,
fever).
• Vaccination in childhood e.g. pneumococcus, meningococcus.
• Good hygiene and nutrition.
2- Genetic counseling.
3- Blood transfusion to keep Hb>8 g/dl + Iron chelation for frequent
transfusions.
4- Folic acid to avoid folate deficiency.
5- Hydroxyurea to enhance production of Hb F, presence of Hb F in the SS
cells decreases polymerization and precipitation of Hb S.

19
Note: Hydroxyurea is cytotoxic and may cause bone marrow suppression it is
indicated in severe cases.
6- Experimental anti-sickling agents.
7- Allogeneic Bone marrow transplant for selected patients.
Homozygous SCA: Hb SS Disease
Treatment of Vaso-Occlusive Crisis
1- Oxygen.
2- Good Hydration (reduces viscosity).
3- Antimicrobials.
4- Correct acidosis if severe.
5- Analgesics/narcotics (give enough to relieve pain).
6- Exchange transfusion for CNS crisis.
25 years old female presented with one week history feature of anemia.
Examination revealed tinge of jaundice and palpable spleen. All the following
are true except
Hb:4g/l,WBC 15X109, 80% neutrophil, retic corrected 16%, DAT positive
for IgG, TSB 4mg/dl peripheral blood film showed spherocytosis.
a-idiopathic cause is the main cause of hemolysis
b-connective tissue disorder need to exclude
c-destruction mainly by phagocytosis in the liver
d-antibody or complement may coated the RBC
e-blood transfusion may be a cause of spherocytosis
20
AUTOIMMUNE HEMOLYTIC ANEMIA
Types:
1- Warm Antibody type :usually IgG.
2- Cold Antibody type: usually IgM.
AUTOIMMUNE HEMOLYTIC ANEMIA (Warm Antibody type)
Pathophysiology: RBC are coated with IgG or complement (C3d) or both
leading to extravascular hemolysis in RES (mainly spleen).
Classification:
1- idiopathic
2- secondary to
• Lymphoproliferative disorders (CLL, Hodgkin’s disease,
Non - Hodgkin’s lymphoma)
• Autoimmune (SLE)
3- Drug induced (penicillin, quinine/quinidine, alpha methyl dopa)
Clinical Features:
Usually insidious :anemia, jaundice and splenomegaly.

21
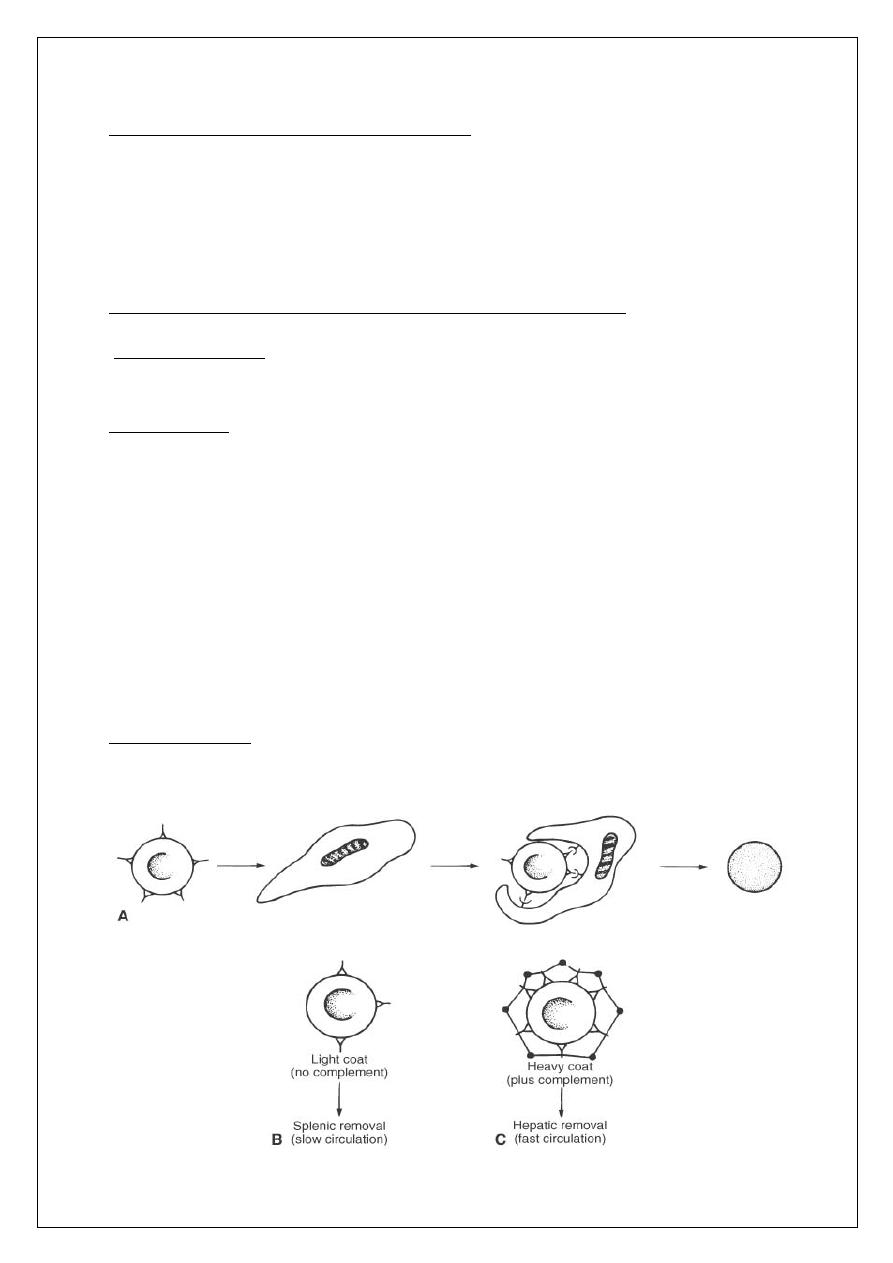
Mechanism of extravascular hemolysis in autoimmune hemolytic anemia. (A)
Macrophage encounters an lgG-coated erythrocyte and binds to it via its Fc
receptors. Thus entrapped, the RBC loses bits of its membrane as a result of
digestion by the macrophage. The discoid erythrocyte transforms into a
sphere. (B) RBC lightly coated with lgG (and therefore incapable of activating
the complement cascade) is preferentially removed in the sluggish circulation
of the spleen. (C) RBC with a heavy coat of lgG; thus, C3b (black circles) can
be removed both by the spleen and the liver
AUTOIMMUNE HEMOLYTIC ANEMIA (Warm Antibody type)
Diagnosis
1- Spherocytes in blood film, reticulocytosis.
2- Indirect hyperbilirubinemia, raised s.LDH.
3- Positive direct antiglobulin test (direct Coombs’) best detected at 37ºC
(hence “warm-reacting antibodies”)
4- Exclude delayed transfusion reaction.
Management
q
Treat underlying cause
q
Corticosteroids: prednisolone 1mg/Kg until response then taper over 2-
3 months.
q
Splenectomy for relapsed and corticosteroid resistant cases .
q
Immunosuppressives like azathioprine for relapses after splenectomy
q
Blood transfusion is used with caution.
q
Add folic acid.

22
Autoimmune Hemolytic Anemia with Cold-Reacting Antibodies
Pathophysiology
q
Either monoclonal or polyclonal IgM Antibodies attach to RBC surface
antigens in peripheral circulation where T < 37ºC.
q
Antibodies will detach from the surface antigen if T > thermal
amplitude
q
Thermal amplitude is the temperature at which IgM is attached to RBC
surface
q
Associated with intravascular hemolysis
Classification
1- Idiopathic
2- Secondary to
• Lymphoproliferative disorders (CLL, Hodgkin’s disease,
non-Hodgkin’s lymphoma)
• Infections (Mycoplasma pneumoniae, EBV).
Clinical Features:
Anemia, acrocyanosis, joint pain, vasculitic rash, Raynaud phenomena, and
rarely splenomegaly.
COLD AGGLUTININ
23
Autoimmune Hemolytic Anemia with Cold-Reacting
Antibodies (IgM)
Diagnosis
1- Anemia, mild reticulocytosis, RBC agglutination in blood film .
2- Positive cold agglutinin test best at 4ºC.
3- Positive direct Coombs’ for complement at any temperature.
Management
1- Treat underlying cause
2- Warm the patient above the thermal amplitude of the antibody
3- Plasmapheresis.
4- Immunosuppressives like chlorambucil.
Non Immune Hemolysis
1- Infections:
Bacterial, Malaria, Babesia.
2- Mechanical:
1- Microangiopathic Hemolysis (MAHA): TTP, HUS, DIC.

24
2- March Hemoglobinurea.
3- Mechanical Cardiac Valves.
3- Snake bite.
4- Burns.
Drug Induced Hemolysis
1- Hapten Mechanism: high dose Penicillin
2- Complement Fixation : quinidine , phenacetin.
3- Autoantibody production: L-dopa, methyldopa.
4- Nonspecific: cephalothin.
5- Metabolic: sulfa drugs.
Hypersplenism
It is a state of sequestration of one or more of blood elements in an
enlarged spleen.
Causes
1- Portal hypertension
2- Myeloproliferative disorders.
3- Thalassemia major.
4- Others.
Diagnosis
1- Reduction of one or more of blood elements.
2- Normal cellularity of bone marrow.
Treatment

25
Treat the underlying condition, splenectomy may be indicated for
increased transfusion requirements.
in Hypersplenism ,the following are:
a-Portal hypertension may be a cause
b- there may be a reduction only in platelet
c- hypocellularity of bone marrow.
d-splenectomy may be indicated for sever cytopenia
E- Howell-jolly body may seen in peripheral blood
