Inherited cardiomyopathies
BMJ 21 November 2011د. حسين محمد جمعه
اختصاصي الامراض الباطنة
البورد العربي
كلية طب الموصل
2011
Inherited cardiac conditions include primary electrical,myocardial, and structural heart diseases, in addition to vascular conditions. The presentation, diagnosis, and management of the different categories of inherited cardiac disease vary greatly. In this review we focus on inherited heart muscle disorders, known as cardiomyopathies. Collectively, they affect about one in 390 people (table⇓).1 However, the true prevalence of each condition
is difficult to determine because evidence of disease may be subtle in asymptomatic patients.
Cardiomyopathies can lead to sudden death as a result of fatal ventricular arrhythmias.
Disease of the heart muscle may also progress with age and lead to heart failure.Early diagnosis and modification of risk factors for premature death may prevent such outcomes and are important.
The diagnostic process has improved in recent years, with advances in clinical cardiological evaluation, and particularly genetic testing.
We review the diagnosis and management of patients with cardiomyopathy for non-specialist doctors and discuss the value of genetic testing in the management of affected families.
This article is based mainly on the findings of medium and large cohort studies,which form the evidence base of the consensus statements of professional bodies.
Which inherited cardiomyopathies might
general clinicians encounter?
On the basis of international estimates (table), an average primary care practice in the United Kingdom of 7380 patients would include about 15 patients with hypertrophic cardiomyopathy, the most common cardiomyopathy.
Patients with less common conditions are likely to cluster in practices with affected families.
Hypertrophic cardiomyopathy is defined as unexplained cardiac hypertrophy, and it often shows an asymmetric pattern—affecting mainly the interventricular septum.
Arrhythmogenic right ventricular cardiomyopathy causes replacement of myocardium with fibro-fatty tissue, particularly in the right ventricle, although the left ventricle may also be affected.
Electrophysiological changes, including arrhythmias, can often pre-date overt structural and functional changes.
Dilated cardiomyopathy is characterised by dilation and impaired contraction of the left or both ventricles, and it is hereditary in about a quarter of cases.
End stage arrhythmogenic right ventricular cardiomyopathy and hypertrophic cardiomyopathy may be difficult to distinguish from dilated cardiomyopathy because left ventricular systolic dysfunction may complicate diagnosis. Different cardiomyopathic features can overlap in individual families, as seen in our patient case study.
What is the role of the family doctor?
Symptomatic patientsPatients may present to their primary care doctor with symptoms caused by myocardial ischaemia, arrhythmia, or heart failure, which can include chest pain, syncope, dyspnoea, palpitations,or stroke. However, a pilot study of the feasibility of heart screening for sudden cardiac arrest in healthy children found that history and physical examination alone are poor predictors of disease in young people, although it is still important to take a family history.
Initial investigation with electrocardiography, with a view to requesting cardiac imaging in the face of an abnormality, is warranted in any patient with worrying symptoms. “Red flag” symptoms (box) include palpitations and exertional dyspnoea accompanied by syncope or seizures in ayoung person.
After a definitive diagnosis, family doctors are often the first point of contact when patients deteriorate. Cardiomyopathies are progressive disorders and complications are a risk as patients age. Any worsening in symptoms of heart failure, signs of ischaemia, or unheralded syncope warrants urgent specialist re-evaluation.
Asymptomatic case finding
Electrocardiographic abnormalities suggestive ofcardiomyopathy may be identified incidentally, such as during preoperative risk evaluation or personal health assessment, or as a result of population based screening programmes that are available in some countries.
Summary points
Primary care doctors play a key role in identifying those at risk of inherited cardiomyopathy .
People who present with symptoms of heart disease, relatives of an affected person, those with a family history of premature sudden death, and those with incidental electrocardiographic abnormalities are at risk of cardiomyopathy .
Most cardiomyopathies are inherited in an autosomal dominant manner Disease severity varies owing to variable penetrance. Specialists should evaluate suspected cases, and pathways to specialist care should be tailored to local healthcare systems
Mutation analysis allows accurate identification of carriers, but diagnosis and risk prediction still rely on clinical markers.
A patient’s perspective
In November 2008, I collapsed while playing tennis at a sports club. I was resuscitated by a quick thinking attendant with a defibrillator.I spent a month at three local hospitals before eventually having a cardiac defibrillator implanted at St George’s Hospital, just before my 50th
birthday.
The members of my family were tested and my sons were found to have a similar heart condition. They are 20 and 16, and have both been fitted with a defibrillator as a precaution. I also have a younger brother who survived a similar collapse around 25 years ago. This was largely unexplained at the time, but he has now been fitted with a defibrillator.
The diagnosis has been uncertain for all concerned.
I had a full range of tests while in hospital and my family has had the same. We feel protected by the cardiac defibrillators without knowing clearly what the underlying problem is.
My defibrillator went off in my first year when I was walking in the Lake District but it was found to be an inappropriate shock, the settings
were altered, and I’ve had no incidents since. At the time it was reassuring to have immediate contact with the pacing clinic at St George’s,
to download the defibrillator data by telephone, and to pay a quick visit to hospital to sort the problem out.
Our ongoing treatment consists of a six monthly family visit to St George’s, supplemented by remote downloads by telephone from home.
It’s a very “light touch” and doesn’t disrupt our lives too much.
The defibrillators do not have that much impact on our daily lives. My sons have to steer clear of overly physical and competitive exercise.
It is a minor inconvenience when passing through airports, and we check all travel destinations for “defibrillator friendly” hospitals.
But it is a worry. No doubt, as parents, we would worry anyway as our children negotiate their teenage years—late nights out, theme parks,different sports, terms away at college, foreign holidays, gap years, and so on. But the defibrillators add an extra dimension.
Advice for non-specialists: red flag symptoms and signs
Consider priority referral for specialist evaluation in the following situations:• Exertional syncope or seizures
• Exertional chest pain in a young person
• Palpitations associated with impaired consciousness
• Family history of cardiomyopathy
• Family history of premature sudden cardiac death
• Incidental finding of an unexpected electrocardiographic abnormality suggestive of cardiomyopathy
Patients may seek medical advice directly after discovering apositive family history of cardiomyopathy or premature sudden
death, or the family doctor may discover a family history of cardiomyopathy on careful history taking. It is always prudent to refer first degree blood relatives of an affected individual to a specialist for cardiological or genetic evaluation (or both), as
recommended by the PHG Foundation UK working group and international consensus.
Because affected families may cluster in general practices, the family practitioner may be uniquely
placed to discover “at risk” people. Referral of more distant relatives for evaluation depends on patterns of inheritance of the disease and is not routine practice in primary care.
Because death certification may not always identify affected people who are asymptomatic and apparently undiagnosed before a sudden death, particularly those under 40 years, it is important to consider cardiomyopathy as a potential cause of death if a patient reports the sudden and unexplained death of a relative. A recent population based study from Europe found that that relatives are often not referred for evaluation of inherited cardiac conditions after a premature death in the family.
Good communication between primary care doctors and cardiogenetic specialists, and clear patient pathways, are needed to facilitate appropriate evaluation of families and identification of family members at potential risk of inherited cardiomyopathy.
What is the role of the cardiogenetics
specialist centre?Evaluation and management of patients with cardiomyopathy should ideally be undertaken by specialists with appropriate expertise in cardiomyopathies, perhaps at a regional or national centre. We advocate a multidisciplinary approach, with liaison between a cardiovascular genetics service and adult and paediatric cardiologists to ensure that family groups are managed in a coordinated manner.
Although it is usual for genetics services to be planned around the investigation of family units, clinical cardiology services may need to be reorganised to fit this model.
An integrated approach may improve diagnostic
certainty in families where results are equivocal in multiple members. This model also allows family-wide education and counselling on inheritance patterns, diagnosis, and management, thereby promoting informed choices by affected and unaffected members of a family.
How is cardiomyopathy diagnosed?
ElectrocardiographyIn patients clinically affected by cardiomyopathy the resting electrocardiograph is rarely normal. In gene carriers who seem to be clinically unaffected (incomplete gene expression) the electrocardiograph is usually normal. Classically, large voltage QRS complexes with ST depression and deep T wave
inversion (figure; part A⇓) are seen in patients with hypertrophic cardiomyopathy, although non-specific electrocardiographic changes may also be seen and may be the only initial indication of disease.
In patients with arrhythmogenic right ventricular
cardiomyopathy, subtle abnormalities may be evident on electrocardiography (figure; part B) in the right precordial leads (V1-V3). These abnormalities include T wave inversion and broadening of the QRS and the “epsilon wave” (notch in the ST segment), which reflect the predilection of the disease for right ventricular muscle. Changes can be more extensive in severe arrhythmogenic right ventricular cardiomyopathy.Patients with dilated cardiomyopathy may also show non-specific abnormalities on electrocardiography (figure; part C).
Specialist investigations
A cardiologist will request echocardiography or magnetic resonance imaging to make a definitive diagnosis in patients with hypertrophic cardiomyopathy and dilated cardiomyopathy;these investigations can identify characteristic ventricular hypertrophy or dilation and impaired systolic function,respectively. Wide variability in gene expression in arrhythmogenic right ventricular cardiomyopathy has led experts to develop a complex diagnostic scoring system that is used to determine the likelihood of disease .
Excluding other conditions
The cardiologist will need to exclude conditions that may mimic inherited cardiomyopathies because these will require different management strategies from the inherited conditions they resemble. Athlete’s heart (as a result of chronic exercise training)and cardiac changes secondary to hypertension and amyloid may mimic hypertrophic cardiomyopathy, and coronary artery disease and chronic alcohol excess may lead to cardiac muscle changes that mimic dilated cardiomyopathy.
Dilated cardiomyopathies pose a particular difficulty, given that only some are inherited; the familial nature of dilated cardiomyopathy can be established only by reviewing family history, which may require investigation of blood relatives.
What is the approach to management of
people with inherited cardiomyopathies?Management of patients depends on the specific condition.
Strategies are broadly divided into treatment of symptoms,
prevention of disease progression, and reduction in the risk of stroke and sudden death; some treatments span more than one of these categories.
Given the relative rarity of inherited cardiomyopathies, the recommendations from professional bodies for therapeutic interventions and risk stratification have been based on multicentre prospective and retrospective cohort observations.
Small case series underpin recommendations for less well proved treatments.
Reducing sudden death
The risk of sudden death as a result of fatal arrhythmias can be reduced by lifestyle changes tailored to removing triggers. For example, participating in competitive sport is universally discouraged. On the basis of randomised trials, drugs such as β blockers and angiotensin converting enzyme inhibitors are commonly used to reduced the risk of death in patients with dilated cardiomyopathy, but drugs have not been shown to be useful in reducing the risk of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy or hypertrophic cardiomyopathy.Implantable cardioverter defibrillators are the only intervention that has been shown to reduce sudden death in most patients with an inherited cardiomyopathy. Longitudinal observational
and multicentre randomised studies have shown that a previous cardiac arrest or haemodynamically compromising ventricular arrhythmia puts people with cardiomyopathy at high risk of recurrence. Such patients should be considered for a secondary
prevention implantable defibrillator.
Implantable defibrillators may also be offered for primary prevention to those who are at sufficiently high risk of sudden death. Risk is determined by the severity of left ventricular systolic dysfunction and conduction disease for patients with dilated cardiomyopathy, arisk scoring system for those with hypertrophic cardiomyopathy,
and severity of disease and symptoms in patients with arrhythmogenic right ventricular cardiomyopathy.
Management of progression of disease and symptoms
Symptoms of heart failure, including dyspnoea, may be reduced by achieving euvolaemia with diuretics, although we advise caution in hypertrophic cardiomyopathy because of the predominance of diastolic dysfunction.For patients with dilated cardiomyopathy, the findings of multiple large randomised controlled trials support treatment with β blockers, angiotensin converting enzyme inhibitors, angiotensin II receptor blockers,and spironolactone, in addition to cardiac resynchronisation with biventricular pacing, all of which are aimed at slowing progression of disease and reducing mortality.
The effectiveness of these treatments in the other cardiomyopathies is less certain, although they are recommended by international guidelines for all patients with left ventricular systolic dysfunction as a result of end stage disease.
In patients with hypertrophic cardiomyopathy, chest pain caused by ischaemia may be ameliorated by β blockers or verapamil.
Symptoms secondary to outflow tract obstruction may be reduced by the addition of disopyramide. Invasive procedures such as septal alcohol ablation and surgical myomectomy may reduce the outflow tract gradient and reduce symptoms. In patients with arrhythmogenic right ventricular cardiomyopathy, symptomatic ventricular arrhythmias may be reduced by sotalol and amiodarone.
Furthermore, radiofrequency ablation can be a useful adjunct in refractory arrhythmias, although
recurrence as a result of progressive disease is a problem.
A minority of patients with severe symptomatic disease, usually as a result of heart failure, receive heart transplants.
Patients with hypertrophic cardiomyopathy and dilated cardiomyopathy are at increased risk of atrial fibrillation, which is important to recognise because of the concomitant thromboembolic risk.
What is the role of genetic mutation analysis?
Diagnosis
Because genetic testing is less than 100% sensitive, it is recommended only after confirmation of the clinical diagnosis,and even then usually in a limited number of genes that cause hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy. Most inherited cardiomyopathies show a mendelian autosomal dominant inheritance pattern,although less common recessive forms have been described,and dilated cardiomyopathy can also be inherited in an X linked pattern.
To date, more than 20 genes have been implicated
in hypertrophic cardiomyopathy, detectable in 60% of families,whereas mutations in seven genes identify almost half of those affected by arrhythmogenic right ventricular cardiomyopathy.Dilated cardiomyopathy is a heterogeneous disorder, and amyriad genetic abnormalities identified account for only around 30% of familial cases.
Cascade familial testing
Once a definite disease causing mutation is identified in an affected patient, the main diagnostic role for genetic testing is identification of carriers within a pedigree, a process known as “cascade testing.” Family members who carry the same genetic mutation may show different severities of disease, and this is known as variable penetrance. Hence, some patients may remainentirely asymptomatic, with little evidence of disease throughout their lives.
However, as mutation carriers, their children remain at risk of inheriting the disorder and potentially being more severely affected. Despite an autosomal dominant inheritance pattern, the clinical picture may give the appearance of “skipped” generations in a single pedigree. Mutation analysis is useful to allow accurate identification of carriers, and it may be cost effective and reassuring because non-carriers are eliminated from clinical assessment.
Prediction of risk
Risk prediction still depends mainly on clinical assessment for patients with cardiomyopathy. Mutational analysis may affect risk; one specific subgroup increases the risk of suddendeath—mutations in the gene encoding lamin A/C (LMNA),which are associated with dilated cardiomyopathy. Research to assess new markers is under way.
What is the value of tracing relatives?
Assuming an autosomal dominant inheritance, all first degree blood relatives of a proband (the first identified affected member in a family) should undergo cardiogenetic evaluation because they have a 50% chance of being a disease carrier. Case findingby family evaluation can prevent future premature deaths through the timely delivery of treatments that reduce risk.
Remember that the proband may be diagnosed after cardiological investigation, which may occur incidentally or in response to symptoms or a cardiac event, or even at autopsy after sudden
death.
Because of the possibility of age related expression, familial evaluation sometimes requires serial follow-up of young relatives who initially have no evidence of disease. As the natural course of arrhythmogenic right ventricular
cardiomyopathy is poorly documented and late onset hypertrophic cardiomyopathy is increasingly recognised, follow-up may be recommended into middle age or later,
particularly in people who carry a disease causing mutation identified in an affected blood relative.
Ongoing research
Research in inherited cardiomyopathies focuses on many different areas:• The identification of previously undescribed disease causing genes and mutations within them
• The scientific study of the molecular consequences of specific mutations and what they can tell us about the underlying mechanisms of disease
• The study of factors that modify the presentation of inherited cardiac conditions and hence explain variable expression
• The development and assessment of new methods for the diagnosis and stratification of risk
• The development of new treatments that may be tailored to the genetic diagnosis
Table
Table 1| Reported prevalence of inherited cardiomyopathiesCondition Prevalence
Hypertrophic cardiomyopathy 1 in 500 (Europe)
Arrhythmogenic right ventricular cardiomyopathy 1 in 1000 to 1 in 10 000 (Europe)
Dilated cardiomyopathy 36.5 in 100 000 (United States)*
*This estimate of prevalence is based on sparse literature and is thought to be an underestimate; it is calculated on the assumption that about 25% of cases of dilated cardiomyopathy are familial.
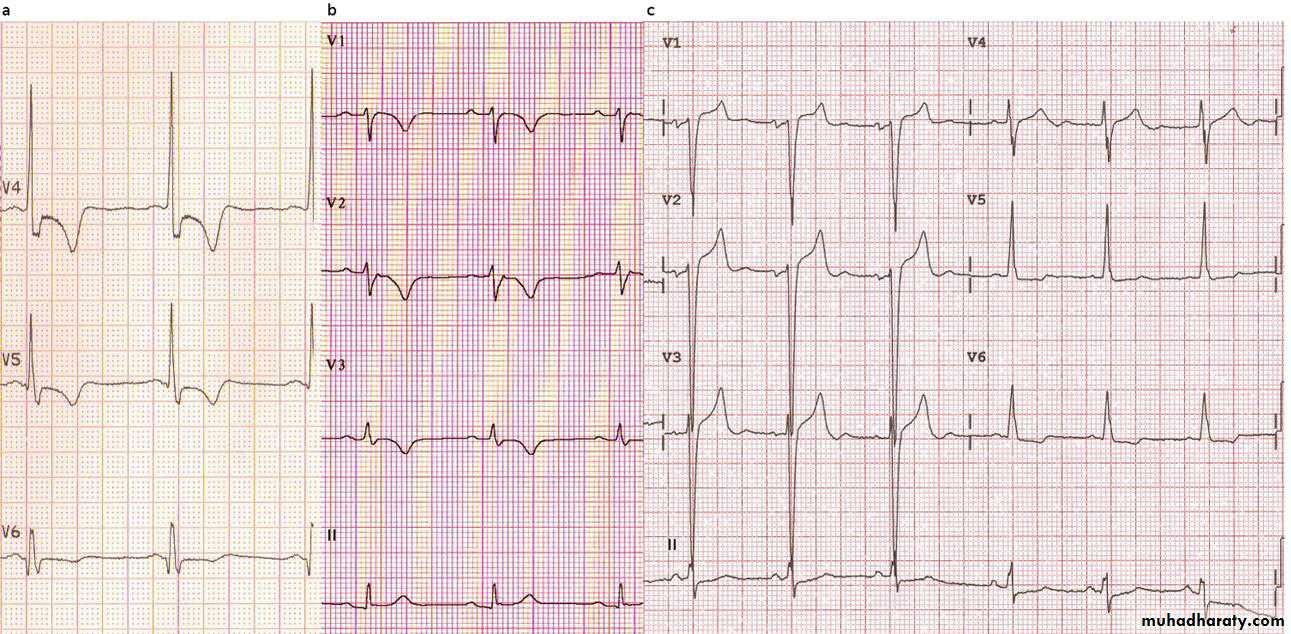
Examples of electrocardiographs from patients with inherited cardiomyopathy.
• Lateral precordial leads of a patient with hypertrophic cardiomyopathy showing Q wave (V6), ST depression, and T wave inversion associated with large voltage QRS complexes.• Right precordial leads of a patient with arrhythmogenic right ventricular cardiomyopathy showing epsilon wave (V2), QRS prolongation, and T wave inversion.
• Precordial leads of a patient with dilated cardiomyopathy showing incomplete left bundle block and left atrial enlargement
BMJ 21 November 2011

