
Endocrine
د. حسين محمد جمعةاختصاصي الامراض الباطنة
البورد العربي
كلية طب الموصل
2011
A 53 year old man attends for a routine check-up. He underwent coronary artery bypass grafting after a myocardial infarction earlier in the year, and seems to be making good progress. He
says he needs to discuss an embarrassing problem. He explains that he has been having erectile dysfunction, which is making him miserable and preventing normal marital relations.
Sexual dysfunction in cardiovascular disease
BMJ 2011
What you should cover
Clarify what the patient means by erectile dysfunction.physical cause is more likely with gradual onset, constant erectile dysfunction with partial or poorly sustained erections, and no full early morning erections.
Check duration of the problem—is it entirely new or
worsening of a pre-existing problem?
Review psychological factors such as performance anxiety,
anxiety about precipitating another coronary event, low mood, stress, and relationship concerns.
Ascertain patient’s main concerns or worries.
Exclude features of hypogonadism such as loss of libido,
loss of body hair, hot flushes, low energy levels,gynaecomastica, and small testicular size.
Review current medications, focusing on those that might
cause erectile dysfunction (for example, β blockers,
thiazides, spironolactone, fibrates, cimetidine,
antidepressants, antipsychotics), or drugs that would contraindicate phosphodiesterase type 5 (PDE5) inhibitors (such as nitrates and nicorandil). Consider any temporal association with onset of erectile dysfunction symptoms.
Review risk factors for sexual dysfunction, such as alcohol
intake, smoking, recreational drug misuse, and weight gain.Review risk factors for or symptoms of other medical conditions (in addition to known cardiovascular disorders) such as diabetes, prostatic disease, depressive illness,
hypothyroidism, or neurological disorders.
What you should do
Physical examination: check blood pressure; examinegenitalia (for small testicular size which may indicate hypogonadism, fibrosis in the shaft of the penis,retractability of foreskin); digital rectal examination of the prostate is indicated in the presence of genitourinary or
protracted secondary ejaculatory symptoms.
Stratify patient’s cardiovascular risk (low,
intermediate/indeterminate, or high risk)
Arrange diagnostic tests—in particular, glucose, cholesterol
(if not measured within previous 12 months), prolactin,
thyroid stimulating hormone, and testosterone levels
between 0800 and 1100. Concentrations of testosterone
may be low owing to illness within the previous 3 months;if this is the case the test should be repeated after 6-8 weeks .Testosterone testing is recommended because deficiency is reversible and can result in PDE5 inhibitors being less effective. Normal values vary, so follow local laboratory guidelines.
Check whether medications may be causing or exacerbating
the problem. Consider alternatives that are less likely tocontribute to erectile dysfunction, such as angiotensin
converting enzyme inhibitors, calcium channel blockers
(except verapamil), loop diuretics, and proton pump
inhibitors. Some evidence indicates that angiotensin
receptor blockers may improve sexual function, and could be the drug of choice for patients with erectile dysfunction who are newly diagnosed with hypertension.
Discuss other potential causes or aggravating factors.
Attention to lifestyle factors and aggressive lipid control can substantially improve erectile dysfunction with or without pharmacotherapy. These factors should already bebeing addressed in patients with known cardiovascular
disease, but awareness of this added benefit may aid compliance.Clarify the patient’s needs, beliefs, concerns, and expectations.
Ask whether the patient has discussed erectile dysfunction with their partner and what the partner’s feelings were.
Openly discuss the lack of high quality evidence to
underpin treatment decisions and the risks and benefits of different approaches. Depending on the underlying reasons
for any particular prescriptions, discuss changing to adifferent medication or a trial of stopping a medication.
Management options
Manage abnormal blood results, including low testosterone, as part of follow-up consultations.In patients stratified at low cardiovascular risk, consider a PDE5 inhibitor.
Use of nitrates or nicorandil is an absolute contraindication
to use of PDE5 inhibitors. Review any nitrateprescription—often patients have not used these drugs since first diagnosis. If they sporadically use glyceryl trinitrate,or carry one “just in case”, then it may be reasonable to prescribe PDE5 inhibitors—ensure that the patient knows to stop glyceryl trinitrate if chest pain develops after taking sildenafil or vardenafil (withhold glyceryl trinitrate for 24 hours) or tadalafil (48 hours), or the consequences can be fatal.
Nitrates are usually prescribed for control of angina symptoms and, unlike calcium channel blockers and β blockers, they have no prognostic benefit, so replacement with other anti-anginals could be discussed.
PDE5 inhibitors are also contraindicated in patients with hypotension (avoid if systolic blood pressure below 90 mm Hg), recent stroke, unstable angina, and recent myocardial infarction (within 6 weeks).
Arrange follow-up to assess therapeutic outcome in terms of erectile response, side effects, and patient’s satisfaction with treatment.Consider stopping a medication if erectile dysfunction is aknown side effect and if a temporal relation exists, after full discussion of uncertainties, risks, and benefits.
Consider referral:
To relationship counselling, sex therapy, or psychology if relevant. These services are not readily available in some areas, in which case empirical treatment may beappropriate.
To urology or other local service (choice of service varies considerably) in patients at low cardiovascular risk and with contraindications to PDE5 inhibitors.
To urology outpatient clinic for those with history of trauma to genital area, pelvis, or spine, or if examination shows an abnormality of penis or testicles. To cardiology if cardiovascular risk is intermediate/indeterminate or high, for further assessment.
• Testosterone Treatment for Young Males
• Testosterone is the primary, though not only, sex hormone produced and used in the male body to effect sexual arousal, performance, and reproductive functions. A healthy testosterone level in young men is normally 700 ng/dl ,and at these levels, muscle and bones will remain strong, body fat is limited, concentration may be increased, mood is normalized, and sexual libido is healthy. Normally prescribed to aging men, testosterone replacement therapy is occasionally needed in younger men because of uncommon medical or hormonal needs.
Testosterone Declines in Young Men
Although older men notice a decline in their testosterone, symptoms in young men need professional diagnosis. Symptoms may include the voice never deepening or other secondary sexual characteristics never developing, such as body hair or the enlargement of penis and scrotum. The clinician will look for incomplete pubertal development or signs of genetic disorders.A low testosterone level for young men is normally recognized as below 400 ng/dl and low testosterone production can be caused by primary testicular failure and pituitary hormone deficiencies. Testosterone levels are apparently decreasing among all young men over the past few decades, according to a recent study in the Journal of Clinical Endocrinology and Metabolism. Anabolic steroid use also reduces natural testosterone production.
Treatment
Treatment options include intramuscular injections, oral tablets, and transdermal preparations, including scrotal and back patches. Oral tablets have minimal effect, as any hormone will be processed by the liver first. Intramuscular injections have been used for some years and can be localized, though the minor pain associated with these injections does deter some patients.Scrotal and back patches transmit the hormone through the skin with varying effect, and some men consider the scrotal patch to be awkward and uncomfortable. Current Sexual Health Reports says that current gel prescriptions show a higher absorption rate of testosterone into the body, but can be dangerous if the man touches a pregnant woman with the same body area.
Risks
Recent studies reviewed in Drug Topics show that testosterone replacement therapies may increase the chances of prostate cancer in men. The study's authors believe that many doctors are prescribing replacement therapy without considering long-range risks and without a true diagnoses of hypogonadism which is an actual defect in the male gonads. Other risks of testosterone treatment in men of any age include acne, cardiovascular disease, testicular atrophy, and benign enlargement of the prostate.Kallman Syndrome
Kallman Syndrome is a rare X-linked recessive disease characterized by reduced or complete absence of the sense of smell (anosmia), underdeveloped genitalia and sterile gonads. It affects primarily males at an incidence of 1 out of 10,000 and the disease becomes apparent when they fail to begin puberty and to develop secondary sexual characteristics.Kallman Syndrome is primarily an X-linked recessive disease affecting mainly males, although there have been rare cases of Kallman Syndrome among females, in which cases the disease was inherited as an autosomal recessive trait. Impaired or lack of sense of smell is caused by the absence of the olfactory bulbs.
Kallman Syndrome also affects the hypothalamus. The hypothalamus produces reduced levels of GnRH, the hormone responsible for the secretion of the hormone LH. LH is the hormone that stimulates gonadal and genital development. In some instances of Kallman Syndrome, GnRH is not produced at all. Decreased or absence of GnRH also causes reduced levels of other hormones including estrogen and testosterone. Patients are therefore at a greater risk for osteoporosis and brittle bone disease. Kallman Syndrome may also be associated with X-linked Ichthyosis and Conradi-Hunermann Syndrome.
Other symptoms also associated with Kallman Syndrome include gynecomastia, bimanual synkinesis (one hand copying the movements of the other hand), shortened fourth metacarpal bone and an absent kidney.
The life span of an affected individual is generally not altered by Kallman Syndrome.
Treatment of this disease involves hormone, estrogen or testosterone, replacement, and pulsatile GnRH or repeated hCG injections. Patients may undergo fertility treatment later in life if they desire to have children.
Couples with a familial history of Kallman Syndrome may reduce their risk of having a child affected with this disease through a new technology called Preimplantation Genetic Diagnosis (PGD). In PGD, embryos are tested for genetic abnormalities and their gender is determined. Only genetically healthy embryos are implanted. Through this mechanism, the chances of having an affected child can be greatly reduced. Due to complex inheritance patterns couples at risk should seek the advice of a genetic counselor to determine whether PGD will be beneficial. PGD can currently test for a variety of other genetic disorders and is continually being improved to include other genetic diseases.
Klinefelter's Syndrome 1 in every 500 to 1000 male births. Instead of the normal XY chromosomes, these individuals have and extra X chromosome making them XXY. Males with Klinefelter's syndrome have two X chromosomes (47-XXY), in rare cases three (48-XXXY) or four (49-XXXXY) X-chromosomes. The X-chromosomes carry genes in terms of development of testicles, sex hormone production and physical sex development in general as well as to a certain extent also height growth. The extra chromosome(s) results in a series of issues
Klinefelter's Syndrome Symptoms
Small Firm TesticlesLow Testosterone
Infertility
Incomplete Masculinization
Female Body Hair Distribution (Sparse facial, armpit, and pubic hair)
Decreased Libido
• Klinefelter's Syndrome Symptoms
• Small Firm Testicles• Low Testosterone
• Infertility
• Incomplete Masculinization
• Female Body Hair Distribution (Sparse facial, armpit, and pubic hair)
• Decreased Libido
• Pictures: XXY Klinefelter Male with Tall Thin Body Shape
• Body Shape Patterns:
• Pear Shaped
• Tall
• Abnormal Proportions (Short Trunk, Long Legs, Long Arms, Lower Body Larger Than Upper)
• Teeth Abnormality - (Taurodontism - Enlarged Pulp and Thin Tooth Surface)
• Photographs: XXY Klinefelter Male with Pear Body Shape, Long Arms and Legs
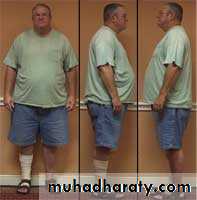
• Varicose Veins, Spider Veins, and Clots
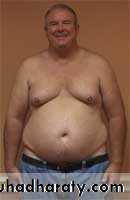
• Gynecomastia Male Breast Deformity• Autoimmune Disorders
• Lupus
• Rheumatoid Arthritis
• Sjogren's Syndrome
• Pictures: XXY Klinefelter Male with Pear Body Shape and Gynecomastia
Pictures: XXY Klinefelter Male Tall Pear Body Shape, Long Arms and Legs
Low EnergyDevelopmental Delays
Difficulty with Motor Skills
Impaired Language / Speech Skills (especially with expressive language)
Learning Disabilities
Normal or High IQ
Social Interaction Difficulties
ADHD (Attention Deficient Hyperactivity Disorder)
Impulse Control Disorder
Depression
Low Self Esteem
Klinefelter syndrome, 47, XXY, or XXY syndrome is a condition in which human males have an extra X chromosome. While females have an XX chromosomal makeup, and males an XY, affected individuals have at least two X chromosomes and at least one Y chromosome.[1] Because of the extra chromosome, individuals with the condition are usually referred to as "XXY Males", or "47, XXY Males".[2]
In humans, Klinefelter syndrome is the most common sex chromosome disorder in males[3] and the second most common condition caused by the presence of extra chromosomes. The condition exists in roughly 1 out of every 1,000 males. One in every 500 males has an extra X chromosome but does not have the syndrome.[4] Other mammals also have the XXY syndrome, including mice.[5]
The principal effects are development of small testicles and reduced fertility. A variety of other physical and behavioral differences and problems are common, though severity varies and many boys and men with the condition have few detectable symptoms.
The syndrome was named after Dr. Harry Klinefelter, who in 1942 worked with Fuller Albright at Massachusetts General Hospital in Boston, Massachusetts and first described it in the same year
Affected males are almost always effectively infertile, although advanced reproductive assistance is sometimes possible. Some degree of language learning impairment may be present, and neuropsychological testing often reveals deficits in executive functionsIn adults, youthful build and facial appearance, or a rounded body type with some degree of gynecomastia .Gynecomastia is present to some extent in about a third of affected individuals, a slightly higher percentage than in the XY population.
The term hypogonadism in XXY symptoms is often misinterpreted to mean "small testicles" or "small penis". In fact, it means decreased testicular hormone/endocrine function. Because of this (primary) hypogonadism, individuals will often have a low serum testosterone level but high serum follicle-stimulating hormone (FSH) and luteinizing hormone (LH) levels. Despite this misunderstanding of the term, however, it is true that XXY men also have microorchidism (i.e. small testicles).[12]
The more severe end of the spectrum of symptom expression is also associated with an increased risk of germ cell tumors, male breast cancer, and osteoporosis, risks shared to varying degrees with females. Additionally, medical literature shows some individual case studies of Klinefelter syndrome coexisting with other disorders, such as pulmonary disease, varicose veins, diabetes mellitus, and rheumatoid arthritis, but possible correlations between Klinefelter and these other conditions are not well characterized or understood.
There are many variances within the XXY population, just as in the most common 46,XY population. While it is possible to characterise 47,XXY males with certain body types, that in itself should not be the method of identification as to whether or not someone has 47,XXY. The only reliable method of identification is karyotype testing.
Diagnosis
A karyotype is used to confirm the diagnosis. In this procedure, a small blood sample is drawn. White blood cells are then separated from the sample, mixed with tissue culture medium, incubated, and checked for chromosomal abnormalities, such as an extra X chromosome.Diagnosis can also be made prenatally via chorionic villus sampling or amniocentesis, tests in which fetal tissue is extracted and the fetal DNA is examined for genetic abnormalities.
Cause
The extra X chromosome is retained because of a nondisjunction event during meiosis (gametogenesis). Nondisjunction occurs with when homologous chromosomes, in the case the X and Y sex chromosomes, fail to separate, producing a sperm with an X and a Y chromosome. Fertilizing a normal (X) egg produces an XXY offspring. The XXY chromosome arrangement is one of the most common genetic variations from the XY karyotype, occurring in about 1 in 500 live male births.Another mechanism for retaining the extra X chromosome is through a nondisjunction event during meiosis II in the female. Nondisjunction will occur when sister chromatids on the sex chromosome, in this case an X and an X, fail to separate. An XX egg is produced which, when fertilized with a Y sperm, yields XXY offspring.
In mammals with more than one X chromosome, the genes on all but one X chromosome are not expressed; this is known as X inactivation. This happens in XXY males as well as normal XX females.
However, in XXY males, a few genes located in the pseudoautosomal regions of their X chromosomes, have corresponding genes on their Y chromosome and are capable of being expressed.[17] These triploid genes in XXY males may be responsible for symptoms associated with Klinefelter syndrome.
The first published report of a man with a 47,XXY karyotype was by Patricia A. Jacobs and Dr. J.A. Strong at Western General Hospital in Edinburgh, Scotland in 1959. This karyotype was found in a 24-year-old man who had signs of Klinefelter syndrome.
Variations
The 48, XXYY (male) syndrome occurs in 1 in 18,000–40,000 births and has traditionally been considered to be a variation of Klinefelter syndrome. XXYY tetrasomy is no longer generally considered a variation of KS,although it has not yet been assigned an ICD-10 code.Males with Klinefelter syndrome may have a mosaic 47,XXY/46,XY constitutional karyotype and varying degrees of spermatogenic failure. Mosaicism 47,XXY/46,XX with clinical features suggestive of Klinefelter syndrome is very rare. Thus far, only about 10 cases have been described in literature.
Treatment
The genetic variation is irreversible. Testosterone treatment is an option for some individuals who desire a more masculine appearance and identity. Often individuals that have noticeable breast tissue or hypogonadism experience depression and/or social anxiety because they are outside of social norms. This is academically referred to as psychosocial morbidity.By 2010 over 100 successful pregnancies have been reported using IVF technology with surgically removed sperm material from men with Klinefelter syndrome
Male Menopause
Male menopause is an informal term used for a condition caused when testosterone levels decrease in aging men. Experts disagree on how widespread the condition is. Some say only around 2 percent of men older than 60 have below-normal testosterone levels. Others say 40 to 80 percent of men older than 70 have it. The Endocrine Society (ES) estimates that millions of American men don’t produce enough testosterone.What is testosterone?
Testosterone is the male sex hormone responsible for male characteristics. It triggers the penis and testicles to form in a fetus. At puberty, it causes the penis and testicles to grow larger. It also causes facial and pubic hair to grow, the voice to deepen, and muscle mass and strength to increase. It governs where fat is distributed on the body and oversees the increase in body height that occurs during adolescence. Testosterone also controls sex drive and sperm production.Testosterone is made by the testicles. A small amount also is made by the adrenal glands. (In women, small amounts of testosterone are made by the ovaries.)
Testosterone production is controlled by gonadotropin-releasing hormone (GnRH),pituitary gland makes luteinizing hormone (LH), which signals the testicles to make testosterone. When there is enough testosterone, the hypothalamus sends a message to the pituitary glad to stop making LH, and the testicles slow down production of testosterone. In an adult male, about
7 mg of testosterone is made each day.
Testosterone production reaches its peak during adolescence and young adulthood. The range of what is considered a “normal” level of testosterone varies widely. Testosterone levels usually decline as a man ages but never drop to zero, as estrogen does for a woman when she reaches menopause.
Klinefelter's syndrome
HemochromatosisKallmann's syndrome
Prader-Willi syndrome
Myotonic dystrophy
Trauma to the testicles
mumps
Radiation treatment or chemotherapy
Treatment of tumors of the testicles
Tumors of the pituitary gland
narcotic pain, prednisone and anabolic steroids
HIV/AIDS
tuberculosis, fungal infection and autoimmune disease that affects the pituitary gland
Aging “andropause.” Andropause usually occurs between the ages of 40 and 55. Beginning around the age of 40, testosterone production decreases by about 1 percent each year.Causes of low testosterone
The normal decline in testosterone because of aging causes some men's hormone levels to go down more than others.
Progressive decrease in muscle mass
Decrease desire in sex (libido)
Erectile dysfunction (ED)
Feeling fat/weight gain
Problems sleeping
Feeling irritable or angry
Loss of motivation
Loss of drive at work
Nervousness
Problems with memory and concentration
Indecisiveness
Lower self-confidence
Tiredness
Depression
Mood swings
Loss of energy
Bone loss
Symptoms of low testosterone
Diagnosis of low testosterone
A simple blood test can determine your testosterone level.. This test should be done in the morning, (7 – 10 AM) when the testicles usually release more testosterone. Normal results usually range from 300 to 1000 ng/dL (or 10 to 38 nmol/L). you may need several tests taken over time.A total testosterone level that is repeatedly less than 250 ng/dL (8.5 nmol/L) indicates a very low level of testosterone production.
Treatment
About 98 percent of the testosterone carried in the blood is bound to two proteins and is not available to the tissues of the body.
The remaining 2 percent, which circulates freely, causes the effects on body tissues. As men age, more testosterone is bound by proteins. Too little free testosterone can cause the symptoms listed above.
For hypogonadism, TRT works to restore sex drive, fertility, muscle strength, and prevent bone loss. But the long-term benefits and risks of TRT in healthy, aging men have not been studied.
In the majority of short-term studies, TRT decreased a man’s fat mass, increased muscle mass, improved strength and increased bone mineral density. Because ED usually has causes other than low testosterone, TRT does not help in the majority of ED cases.
High doses of testosterone can cause sleep problems and infertility, and increase the risk for stroke, benign growth of the prostate gland. If an undetected prostate cancer is present, experts worry that TRT could cause rapid growth of the cancer. Enlarged breasts and acne can also be side effects of TRT.
TRT can be given by injections every two to three weeks; by skin patches; and by implanted pellets that are inserted every four to six months. A recently approved form of testosterone is by Patches Gel Tablets (stick to the gums).
Testosterone is not given orally because it can affect the liver, raise total cholesterol levels, and decrease high density lipid.
Possible Risks of Testosterone Treatment:
A high red blood cell count. Occasional stopping of breathing during sleep (sleep apnea) An increase in prostate enlargement or prostate cancer growth . all men over 50 years of age should be carefully monitored for prostate cancer during testosterone treatment. Men with breast cancer or known or suspected prostate cancer should not receive testosterone therapy.
Testosterone Preparations Available in the United States
Depot esters (Depo-Testosterone, Delatestryl)Genital skin patch (Testoderm)
Nongenital skin patch (Androderm)
Gel (AndroGel, Testim)
Buccal (Striant)
Male Hypogonadism
Definition and prevalence
Male hypogonadism is defined as the failure of the testes to produce androgen, sperm, or both. Although the disorder is exceedingly common, its exact prevalence is uncertain.
Testosterone production declines with advancing age; 20% of men older than 60 years and 30% to 40% of men older than 80 years have serum testosterone levels that would be subnormal in their younger adult male counterparts.
This apparent physiologic decline in circulating androgen levels is compounded in frequency by permanent disorders of the hypothalamic-pituitary-gonadal axis .These include the transient deficiency states associated with acute stressful illnesses, such as surgery and myocardial infarction, and the more chronic deficiency states associated with wasting illnesses, such as cancer and acquired immunodeficiency syndrome.
Male factor infertility is probably responsible for one third of the 10% to 15% of couples who are unable to conceive within 1 year of unprotected intercourse. Most of these male-associated cases result from diminished, absent, or faulty spermatogenesis. In addition to abnormal sperm production, other conditions, including obstructive ductal disease, epididymal hostility, immunologic disorders, and erectile or ejaculatory dysfunction should be considered.
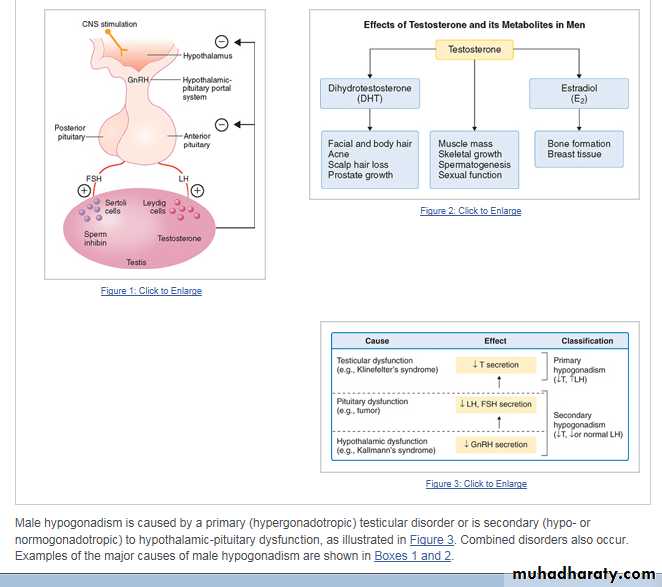
Pathophysiology
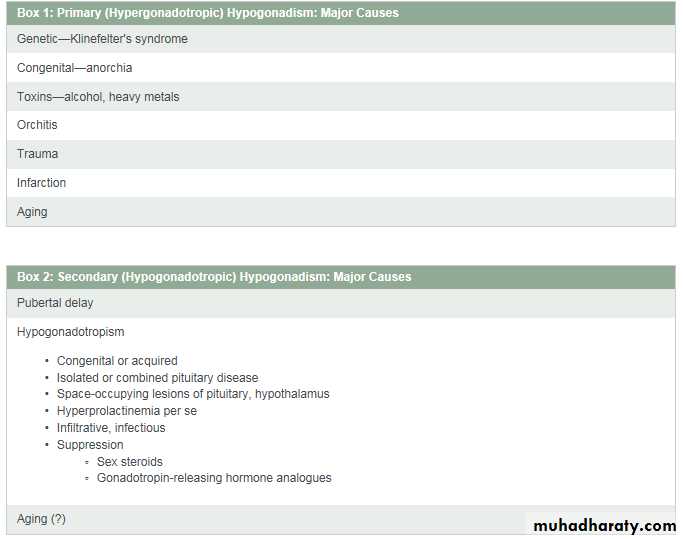
The physiologic regulation of the hypothalamic-pituitary-gonadal axis is shown in Figure 1. Circulating testosterone is largely protein-bound—the major protein is sex hormone–binding globulin (SHBG)—with only 2% present as the biologically active or free fraction. Some clinicians believe that the bioavailable fraction, the fraction present in the supernatant after ammonium sulfate precipitation, representing testosterone loosely bound predominantly to serum albumin, is more meaningful.Hepatic SHBG production rises with aging and thyroid hormone excess and declines in hyperinsulinemic states (obesity and type 2 diabetes), so that free testosterone values may not always be concordant with total testosterone values. The biologic effects of testosterone may be mediated directly by testosterone or by its metabolites 5a-dihydrotestosterone or estradiol (Fig. 2).
Signs and symptoms
Birth and InfancyPersistent failure of the testes to descend may be an early manifestation of testicular dysfunction. In addition, a normally formed but hypotrophic penis may provide a clue to an abnormality of the hypothalamic-pituitary-gonadal axis.
Puberty
Delayed, arrested, or absent testicular growth and secondary sexual characteristic development are hallmarks of pubertal disorders. Skeletal proportions may be abnormal (eunuchoid) with more than a 5-cm difference between span and height and between pubis-floor and pubis-vertex dimensions.
Adulthood
Manifestations in adults are generally more subtle. Perhaps the minor contribution of adrenal androgens (or androgenic precursors) may substitute for testicular deficiency once the target tissues have been fully developed. Moreover, ingrained behavior patterns may be resistant to androgenic hormone deficiency. Certainly, prolactin excess, testosterone deficiency, or both in men may result in impaired libido and erectile dysfunction.The yield of finding hyperprolactinemia or testosterone deficiency, or both, in patients presenting with these symptoms is generally considered to be low, usually less than 5%. However, a large survey of patients with erectile dysfunction presenting to a Veterans Affairs center has suggested that the prevalence of these abnormalities is substantial—18.7% of patients with low testosterone levels and 4.6% with elevated prolactin levels. 1
The first manifestation of hypogonadism may be a consequence of a large space-occupying intrasellar or parasellar lesion manifested by headaches, bitemporal hemianopia, or extraocular muscle palsy. Galactorrhea as a manifestation of hyperprolactinemia is rare, but rarely sought. Unexplained osteoporosis or mild anemia sometimes is the clue to an underlying hypogonadal state. Some common clinical conditions associated with male hypogonadism are listed in Box 3. The subject of androgen deficiency and the aging man is dealt with in greater detail later in this chapter.
Because the acute effect of stressful illness may result in a transient lowering of testosterone levels, a confirmatory early morning specimen should be obtained. Measurement of free testosterone levels or bioavailable testosterone levels, determined adequately in select commercial laboratories, may provide additional information (see later, “Pathophysiology”).
For example, free testosterone levels may be lower than expected from the total testosterone level as a result of aging and higher than expected in insulin-resistant individuals, such as in obesity. In addition, serum follicle-stimulating hormone (FSH), luteinizing hormone (LH), and prolactin levels should be determined to help delineate the cause of the testosterone-deficient state.
If gonadotropin levels are not elevated, despite clearly subnormal testosterone values, anterior pituitary (thyroid-adrenal) function should be determined by measuring free thyroxine and thyroid-stimulating hormone levels, as well as an early morning cortisol level. A magnetic resonance imaging (MRI) scan of the brain and sella should be considered. An exception to this recommendation is the condition of morbid obesity, in which both total and free testosterone levels are typically low and gonadotropin values not elevated.
Hyperprolactinemia, even of a small degree, may also warrant ordering MRI, because interference of hypothalamic-pituitary vascular flow by space-occupying, stalk-compressing lesions will lead to disruption of the tonic inhibitory influence of hypothalamic dopamine, and result in modest hyperprolactinemia (20- to 50-ng/mL range).
A semen analysis should be performed when fertility is in question.
Testosterone Replacement ,In addition to monitoring testosterone levels periodically, prostate screening and measurement of hemoglobin and hematocrit levels must also be performed at intervals when the patient is on therapy.
Prostate Screening
Levels of prostate-specific antigen (PSA) should be checked at 3, 6, and 12 months. If the patient is truly hypogonadal to begin with, expect a significant rise at the 3-month assessment. Thereafter, the usual criteria apply regarding the possible presence of an underlying malignancy (higher than 4 ng/mL, or rate of increase more than 1.5 ng/mL/2 yr or more than 2 ng/mL overall).These criteria continue to be revised by our urology colleagues, tending to become more stringent with time. For example, a PSA rise of more than 1 ng/mL/yr has been suggested as an early warning guide, and closer surveillance has been recommended, even at rates of 0.7 to 0.9 ng/mL/yr. 2A digital rectal examination should be performed at 3 to 6 months and at 1 year after therapy is initiated. A urologic consultation should be obtained if indicated.
Hemoglobin and Hematocrit Levels
Hemoglobin (Hb) and hematocrit (Hct) levels should be checked periodically. Incremental increases are to be expected, but an Hb level higher than 17.5 g/dL, Hct higher than 55%, or both suggests overtreatment, occasionally abuse. Greater increments tend to occur more frequently with the intramuscular than with the transdermal preparations. If dosage adjustments do not solve the problem, look for another underlying cause.Contraindications
Physicians should take into consideration a number of clinical situations in which absolute or relative contraindications for the use of testosterone exist (Box 5). It should be noted that no long-term studies in large numbers of patients (neither young or old) have been performed, so potential risks and benefits need to be individualized.Box 5: Contraindications for Testosterone Replacement
• Breast carcinoma (history or presence)• Prostate carcinoma (history or presence)
• Severe benign prostatic hyperplasia
• Abnormal digital rectal examinations
• Elevated levels of prostate-specific antigen
• Age (no limit established; possibly older than 80 years)
• Psychopathology
• Sleep apnea (potential for worsening)
• Hypercoagulable states
• Polycythemia (hematocrit > 51%)
Box 6: Potential Benefits of Testosterone Therapy
Body composition Increase in lean body mass
Decrease in fat mass
Bone Increased bone density
No fracture data available
Mood, well-being
Sexual function
Cognitive function
Muscle strength, physical function
Older Men
The aging man represents a special case and has been the subject of a review. 4There is a well-known decline in testosterone production with aging in otherwise healthy men. This decline in mean values can be seen in free testosterone levels, beginning in the mid-40s (some clinicians suggest even earlier), as a consequence of increasing SHBG levels, mechanism unknown. Total testosterone levels decline on average beyond 70 years.The diurnal rhythm, seen in younger men, is lost beyond 60 years. 5Although testicular volume also declines in this age group, spermatogenesis may be well maintained into the 80s or even beyond. Gonadotropin levels tend to rise after 70 years, indicating that the testosterone deficiency is usually primary. 6Figure 4 schematically presents these hormonal changes with age. Using the criterion of a low testosterone value, and remembering that there is considerable variability in commercially available tests regarding normal young adult ranges, it has been estimated that 7% of 40- to 60-year-olds, 22% of 60- to 80-year-olds, and 36% of 80- to 100-year-olds are hypogonadal. 7
National guidelines
The American Association of Clinical Endocrinologists has published 2002 updated guidelines for the evaluation and treatment of hypogonadism in adult male patients. 11This review, geared particularly for endocrinologists, expands on some of the areas reviewed in this chapter and provides a more detailed look into aspects of male infertility.The Endocrine Society has published clinical practice guidelines 12for testosterone replacement therapy. The major recommendations are summarized in Box 7.

Summary
Male hypogonadism is defined as the failure of the testes to produce androgen, sperm, or both.Signs and symptoms vary according to age.
Diagnosis requires the determination of low testosterone levels. Normal ranges vary among laboratories. Measurement of free testosterone levels or bioavailable testosterone levels may provide additional information, in addition to serum follicle-stimulating hormone, luteinizing hormone, and prolactin levels. MRI scans of the brain and sella should be considered.
Androgen replacement therapy is used for the treatment of male hypogonadism. In addition to monitoring testosterone levels periodically, prostate screening by digital rectal examination and prostate specific antigen determinations at periodic intervals when the patient is on therapy should be carried out. Hemoglobin and hematocrit levels should also be checked periodically.
When Should Testosterone Replacement Therapy Be Initiated?
Clinicians should not become too focused on specific numerical thresholds and ranges in defining TD, but instead accept that there are no clear dividing lines between normal and deficient testosterone levels in the blood. The different societies are generally in agreement that• TT levels >350 ng/dL do not require treatment;
• a trial of testosterone therapy can be considered in men with unquestionably low or borderline low testosterone levels and a consistent clinical picture of TD. In the United States, most men initiate testosterone replacement therapy with a topical gel.
Prader-Willi syndrome (PWS)
is a disorder caused by a deletion or disruption of genes in the proximal arm of chromosome 15 or by maternal disomy in the proximal arm of chromosome 15. Commonly associated characteristics of this disorder include diminished fetal activity, obesity, hypotonia, mental retardation, short stature, hypogonadotropic hypogonadism, strabismus, and small hands and feet.In 1887, Langdon-Down described the first patient with Prader-Willi syndrome as an adolescent girl with mental impairment, short stature, hypogonadism, and obesity and attributed these symptoms to polysarcia.[1 ]In 1956, Prader et al reported a series of patients with similar phenotypes.[2 ]In 1981, Ledbetter et al identified microdeletions within chromosome 15 and determined it to be the site for Prader-Willi syndrome.
Pathophysiology
Prader-Willi syndrome is the first human disorder attributed to genomic imprinting. In such disorders, genes are expressed differentially based on the parent of origin. An imprinting center has been identified within 15q11-13; gene expression may be regulated by DNA methylation at cytosine bases.[5 ]Prader-Willi syndrome results from the loss of imprinted genomic material within the paternal 15q11.2-13 locus.Frequency
United States
Most cases of Prader-Willi syndrome are sporadic. Burd et al reported a prevalence rate of 1 per 16,062 population.[15 ]Butler reported a prevalence rate of 1 per 25,000 population.
International
Prader-Willi syndrome has been reported worldwide. Reported prevalence rates for Prader-Willi syndrome range from 1 per 8000 population in rural Sweden to 1 per 16,000 population in western Japan.[17,18 ] Despite findings that suggest a prevalence rate of 1 per 52,000 population in the United Kingdom, Whittington et al estimate that the actual prevalence rate is higher and propose a true prevalence rate of 1 per 45,000 populationMortality/Morbidity
Complications due to obesity (eg, slipped capital femoral epiphyses, sleep apnea, cor pulmonale, type 2 diabetes mellitus) and behavioral problems are major contributors to morbidity and mortality in individuals with Prader-Willi syndrome (see Complications). Lamb et al reported premature development of atherosclerosis with severe coronary artery disease in an patient aged 26 years with Prader-Willi syndrome, morbid obesity, and non–insulin-dependent diabetes mellitus.[20 ]Wharton et al described a series of 6 patients with Prader-Willi syndrome with dramatic acute gastric distention preceded by symptoms of gastroenteritis.[21 ]One half of the cases rapidly progressed to massive gastric dilatation and gastric necrosis. One patient died of overwhelming sepsis and disseminated intravascular coagulation. Gastric dilatation spontaneously resolved in 2 children.
Gastrectomy was performed in 2 patients; in one patient, gastrectomy was subtotal and distal, whereas in the other patient, gastrectomy was combined with partial duodenectomy and pancreatectomy. An autopsy series by Stevenson et al reported gastric rupture and necrosis as the confirmed cause of death in 3% of the patients with Prader-Willi syndrome, with another 4 suspected cases of gastric necrosis.[22 ]
In a series of 152 patients with Prader-Willi syndrome, choking episodes were reported as the cause of death in 7.9%.[23 ]Another series of patients noted 8 children and 2 adults who had unexpected death, with small adrenal glands noted in 3 of 8 children, raising suspicion for underlying adrenal insufficiency.[24 ]
Race
Differences in prevalence rates between racial groups have not been consistently reported. However, in a study of 10 blacks with Prader-Willi syndrome, Hudgins et al (1998) suggested that clinical features in black patients differ from those of white patients.[25 ]In black patients, growth is less affected, hand lengths are usually normal, and the facies are less typical.Sex
Prader-Willi syndrome is caused by the loss of the paternal copy in the proximal arm of chromosome 15 in the region of 15p11-13. Differences in prevalence rates between sexes have not been reported.
Age
Prader-Willi syndrome is a genetic disorder with lifelong implications.
Clinical
History
Infants with Prader-Willi syndrome (PWS) commonly exhibit hypotonia, poor suck (with requirement of gavage feedings), weak cry, and genital hypoplasia (eg, cryptorchidism, scrotal hypoplasia, clitoral hypoplasia). Neonatal hypotonia is one of the hallmark features of this disorder and is a valuable clue to initiate diagnostic testing.
Toddlers with Prader-Willi syndrome demonstrate late acquisition of major motor milestones (eg, sitting at age 12 mo, walking at age 24 mo).
Children aged 1-6 years present with symptoms of hyperphagia with progressive development of obesity.
Short stature is generally present during childhood; a minority of patients present later with lack of pubertal growth spurt.
Sleep disturbances, ranging from central or obstructive sleep apnea to narcolepsy, are common.[28 ] Exacerbation of obstructive sleep apnea shortly after initiation of growth hormone therapy is a recent concern.
Most patients with Prader-Willi syndrome have growth hormone deficiency, as determined with provocative testing.[27 ]
Pubic and axillary hair may grow prematurely in children with Prader-Willi syndrome, but other features of Prader-Willi syndrome are generally delayed or incomplete.[27 ]
Testicular descent has occurred as late as in adolescence; menarche may occur as late as age 30 years in the presence of significant weight loss.
Patients with Prader-Willi syndrome often exhibit behavioral problems.[29 ]
Young children exhibit temper tantrums, stubbornness, and obsessive-compulsive behaviors.
Behavioral issues often compromise the level of academic performance. Obsessive-compulsive behaviors and perseveration are challenging for children with Prader-Willi syndrome in the classroom setting.[30 ]Features of psychosis are present in 5-10% of young adults with Prader-Willi syndrome.[31 ]
Food-seeking behaviors may include eating garbage, eating frozen food, and stealing resources to obtain food. High thresholds for vomiting and pain tolerance can complicate binging on spoiled foods and delay treatment for GI disease. Death due to choking episodes has been reported.[23 ] After episodes of binge eating (eg, at holidays), both thin and obese individuals with Prader-Willi syndrome have developed abdominal discomfort, with acute gastric dilation observed using radiography. Some patients have developed gastric necrosis.[22 ]
Mild mental retardation is common.
Obesity complications (eg, sleep apnea, cor pulmonale, diabetes mellitus, atherosclerosis), hypogonadism (osteoporosis), and behavioral issues are common problems in adults with Prader-Willi syndrome.[32 ]
Physical
Holm et al established the following diagnostic criteria for Prader-Willi syndrome. Based on these guidelines, the diagnosis of Prader-Willi syndrome is highly likely in children younger than 3 years with 5 points (3 from major criteria) or in those older than 3 years with 8 points (4 from major criteria).Major criteria (1 point each)
CNS - Infantile central hypotoniaGI - Infantile feeding problems and/or failure to thrive
Nutrition - Rapid weight gain in children aged 1-6 years
Craniofacial - Characteristic facial features such as narrow bifrontal diameter, almond-shaped palpebral fissures, narrow nasal bridge, and down-turned mouth
Endocrine - Hypogonadism
Developmental - Developmental delay and/or mental retardation
Minor criteria (one half point each)
Neurologic - Decreased fetal movement and/or infantile lethargyPulmonary - Sleep disturbance and/or sleep apnea
Endocrine - Short stature for predicted height by mid adolescence
Dermatologic - Hypopigmentation
Orthopedic - Small hands and feet
Orthopedic - Narrow hands with straight ulnar border
Ophthalmologic – Esotropia and/or myopia
Dental - Thick viscous saliva
Otolaryngology - Speech articulation defects
Psychiatric - Skin picking (Some patients with Prader-Willi syndrome have become anemic from chronic rectal bleeding secondary to skin picking.)
Supportive criteria (no points)
Neurology - High pain threshold and normal neuromuscular evaluation for hypotonia
Gastroenterology - Decreased vomiting
Endocrinology - Ineffective thermoregulation, early adrenarche, and/or osteoporosis, adrenal insufficiency[27 ]
Orthopedics – Scoliosis or kyphosis[34 ]
Developmental - Jigsaw puzzle proficiency[33 ]
Causes
Prader-Willi syndrome is due to the loss of the paternal copy of chromosome 15q11.2-13.[4 ]Most cases of Prader-Willi syndrome are sporadic. More than 70% of patients have a deletion of the paternal copy; approximately 25% of patients with Prader-Willi syndrome have maternal uniparental disomy in chromosome 15. The remainder of patients with this disorder have a translocation or other structural alteration in chromosome 15.
Most manifestations of Prader-Willi syndrome are attributable to hypothalamic dysfunction
Anxiety Disorder: Obsessive-Compulsive Disorder
ObesityCryptorchidism
Obesity-Hypoventilation Syndrome and Pulmonary Consequences of Obesity
Failure to Thrive
Obstructive Sleep Apnea Syndrome
Fragile X Syndrome
Osteoporosis
Growth Hormone Deficiency
Short Stature
Hypogonadism
Sleep Apnea
Differential Diagnoses
Other Problems to Be Considered
Angelman syndromeScoliosisHypotoniaBardet-Biedl syndromeCohen syndromeAlbright hereditary osteodystrophy
Laboratory Studies
Genetic testingGenetic testing for Prader-Willi syndrome (PWS) includes chromosomal analysis and assessment for methylation patterns in the Prader-Willi syndrome region.
Methylation patterns can be determined with Southern blot hybridization or polymerase chain reaction (PCR) using DNA primers that can detect methylated cytosine.
Analysis for underlying uniparental disomy requires samples from both parents and the child with Prader-Willi syndrome.
Fluorescent in situ hybridization (FISH) can be used to confirm prenatal diagnosis when a deletion in the 15q region is suspected after chorionic villus sampling or amniocentesis.
In a patient with an imprinting center mutation, test both biological parents for the presence of asymptomatic mutations in the imprinting center; such mutations indicate a higher risk for recurrence.
Hypogonadism
Most patients with Prader-Willi syndrome have hypothalamic dysfunction that manifests as short stature, central obesity, hypogonadism, and osteoporosis.Fasting measurements of serum insulinlike growth factor-1 (IGF-1) and insulinlike growth factor binding protein-3 (IGFBP-3) levels are good screening measurements for underlying growth hormone deficiency.
Refer patients with diminished growth velocity and abnormal levels of IGF-1 and IGFBP-3 to a pediatric endocrinologist for provocative growth hormone stimulation testing.
Assess thyroid and adrenal status in patients when clinically warranted.
Hypopituitarism has been reported in some patients with Prader-Willi syndrome.
Obesity
Measure glycosylated hemoglobin inpatients with Prader-Willi syndrome who are obese to assess for the development of type 2 diabetes mellitus as clinically warranted, especially if the patient is taking growth hormone supplementation.Evaluate patients with Prader-Willi syndrome for biochemical evidence of pickwickian syndrome (eg, hypercarbia, polycythemia) as clinically warranted.
If symptoms suggest sleep apnea or narcolepsy, perform a sleep study with multiple sleep latency testing.
Imaging Studies
Individuals with Prader-Willi syndrome are at risk for pathologic fractures secondary to underlying osteoporosis. A high pain tolerance may allow for minimal symptoms of discomfort with obvious deformity. Patients with Prader-Willi syndrome may require the following imaging studies:
MRI of the head (to evaluate for hypopituitarism)
Serial dual energy x-ray absorptiometry (DEXA) scanning (for detection and monitoring of osteoporosis)
Scoliosis films
Chest radiography (if cor pulmonale is suspected)
Other imaging modalities as clinically dictated (eg, extremity film for limp evaluation, hip films to screen for hip dysplasia[40 ])In patients who suddenly develop abdominal distension, abdominal pain, or a decrease in appetite, imaging including plain abdominal radiography, abdominal ultrasonography, or CT scanning and gastrointestinal series may be warranted to screen for possible conditions such as acute gastric dilation, cholelithiasis, or pancreatitis.
Procedures
Assess the growth hormone axis and adrenal axis under the supervision of an endocrinologist if clinically warranted.
Treatment
Medical CarePatients with Prader-Willi syndrome (PWS) frequently require medical care for the following:
Initial management of hypotonia or poor feeding
Evaluation for hypogonadism or hypopituitarism
Management of obesity
Monitoring for scoliosis
Therapy for behavioral issues
On June 20, 2000, the US Food and Drug Administration (FDA) approved the use of growth hormone in children with genetically confirmed Prader-Willi syndrome and evidence of growth failure.
Surgical Care
Patients with Prader-Willi syndrome may require surgical care for treatment of complications of obesity, treatment of cryptorchidism, and scoliosis intervention. They may require urgent surgical attention for abdominal issues. Because of the high pain tolerance and decreased ability to vomit, they may present late with symptoms of cholecystitis, appendicitis, or acute gastric dilation with risk for progression to necrosis.
Tonsillectomy, adenoidectomy, or tracheostomy placement may be required in patients with obstructive sleep apnea.
Biliopancreatic diversion and gastric bypass surgery have been ineffective for long-term weight reduction.Significant disruption in the enterohepatic circulation of bile acids may result in deficiencies of fat-soluble vitamins and steatorrhea with anal pruritus due to bile acids.
Anal pruritus may exacerbate rectal-picking compulsions. Deficiencies of fat-soluble vitamins may exacerbate the following:
Osteoporosis (vitamin D)
Hypochromic anemia (vitamin E)
Hyporeflexia (vitamin E)
Spinocerebellar ataxia (vitamin E)
Coagulopathy (vitamin K)
Night blindness (vitamin A)
Enhanced susceptibility to infections (vitamin A)
Consultations
Patients with Prader-Willi syndrome may require the support of the following specialists:[40 ]Geneticist for initial diagnosis and counseling
Developmental pediatrician for stimulation programs
Endocrinologist for management of hypogonadism
Nutritionist for dietary counseling
Ophthalmologist for management of strabismus
Pulmonologist for management of sleep apnea
Psychiatrist, psychologist, or both for management of behavioral issues
Gastroenterologist for GI issues
Patients with Prader-Willi syndrome have hyperphagia (onset in children aged 1-6 y) and diminished basal metabolic rate. Various treatment modalities for weight control, ranging from behavioral modification to anorexic agents, have been largely unsuccessful in curbing hyperphagia. However, these modalities may yield some success when used at group home settings.
Diet
Significant dietary restrictions are not implemented during early childhood to ensure optimal myelination.
Institution of a balanced hypocaloric diet (1000 calories with supplementation of vitamins and calcium) is generally implemented at early school age with careful monitoring by a dietitian.
As children with Prader-Willi syndrome become ambulatory, limitation of access to foods is essential for modulation of weight. Placement of locks on cupboards and refrigerators, use of smaller dishes, and restriction of access to food in the school environment help deter excessive weight gain.
In patients with morbid obesity, a protein-sparing modified fast with careful medical and nutritional supervision over several weeks may facilitate short-term weight loss.[45 ]
Activity
Patients with Prader-Willi syndrome have hypotonia and require supplemental occupational and physical therapy to promote acquisition of gross and fine motor skills and to strengthen spinal musculature in order to minimize scoliosis.[34 ]Encouragement of physical activity at home, at school (eg, increased physical education periods), and in the community (eg, Special Olympics) is essential for modulation of weight.
Care providers should be instructed in the Heimlich maneuver.
Medication
Currently, no medications have been found to effectively modify hyperphagia. Growth hormone therapy in patients with growth hormone deficiency improves lean body mass, corrects osteopenia, does not appear to enhance the development of scoliosis, and anecdotally modulates behavior in some patients. Supplementation of sex steroids does improve secondary sex characteristics but may aggravate behavioral disorders.[27 ]Growth hormone agents
improve symptoms of growth hormone deficiency.
Human growth hormone (Saizen, Genotropin, Humatrope, Nutropin, Serostim)
Stimulates growth of linear bone, skeletal muscle, and organs. Stimulates erythropoietin, increasing RBC mass.Dosing
Adult
Not established
Pediatric
0.15-0.3 mg/kg/wk SC initially; divide into daily or 6 times/wk subcutaneous injections; adjust dose to effect
Interactions
Glucocorticoids may decrease growth-promoting effects
Contraindications
Documented hypersensitivity; closed epiphyses; actively growing intracranial tumor; any underlying intracranial lesion
Precautions
PregnancyC - Fetal risk revealed in studies in animals but not established or not studied in humans; may use if benefits outweigh risk to fetus
Precautions
Caution in diabetes; reconstitute with sterile water for injection if administering to newborns
Follow-up
Further Inpatient CarePatients with Prader-Willi syndrome (PWS) may require inpatient evaluation and treatment for hypotonia and poor feeding during infancy.
Individuals with Prader-Willi syndrome and other medical issues, including scoliosis and complications of obesity or pickwickian syndrome, may require inpatient therapy.
Patients with severe behavioral problems may merit admission to a facility staffed with individuals with long-term experience with Prader-Willi syndrome.
Further Outpatient Care
Further outpatient care is targeted toward management of hypogonadism, obesity, and behavioral issues.Transfer
Patients with Prader-Willi syndrome and significant behavioral issues recalcitrant to traditional therapies may benefit from transfer to a center, such as the Children's Institute in Pittsburgh, staffed with individuals with experience in treatment of people with Prader-Willi syndrome.
Deterrence/Prevention
Patients with Prader-Willi syndrome have hyperphagia and require restricted access to foods to minimize weight gain. Binge-eating episodes may predispose patients to development of food poisoning and acute gastric dilation. Caregivers of patients with Prader-Willi syndrome should be instructed in the Heimlich maneuver.
Evaluate males with Prader-Willi syndrome and cryptorchidism for gonadotropin-releasing hormone (GnRH) and orchiopexy.[27 ]
Routinely monitor for symptoms of sleep apnea. Obtain a sleep study within the few months after initiation of growth hormone therapy at the first sign of symptoms.[41 ]
Routinely screen children for scoliosis.[34,40 ]
Regularly screen patients with obesity for evidence of type 2 diabetes mellitus.[27 ]
Screen for thyroid and adrenal function when indicated.[40 ]
Complications
Patients with Prader-Willi syndrome can develop complications from the following:Hypogonadism (osteoporosis/pathologic fractures)
Obesity due to hyperphagia and hypometabolism (secondary to hypopituitarism): This predisposes patients to premature death from cardiorespiratory failure.
Slipped capital femoral epiphyses/hip dysplasia
Sleep apnea: Patients with Prader-Willi syndrome have a primary disturbance in central control of the respiratory drive with diminished responsiveness to hypercapnia during quiet sleep.
Cor pulmonale
Type 2 diabetes mellitus
Neoplasias (eg, Wilms tumors,[46 ]testicular neoplasias,[47 ] multiple endocrine neoplasia [MEN1],[48 ]neoplasias, hematologic neoplasias [leukemia][49 ]): Various neoplasias have been rarely reported in patients with Prader-Willi syndrome.
Binge-eating episodes: These episodes may predispose patients to choking episodes (which require the Heimlich maneuver), acute gastric dilation with risk of gastric necrosis, and food poisoning from consumption of contaminated food.
Prognosis
Patients with Prader-Willi syndrome frequently reach adulthood and are able to function in a group home setting, performing vocational work or attending community college classes.
Diminished sensitivity to pain and diminished capacity to vomit may delay the diagnosis of underlying disease (eg, appendicitis).
Complications from hypogonadism (eg, osteoporosis/pathologic fracture), behavioral issues (eg, temper tantrums, stubbornness, psychoses), and morbid obesity (eg, type 2 diabetes mellitus, cor pulmonale) may shorten life expectancy and may affect the quality of life.
Patients with Prader-Willi syndrome can be mainstreamed into the classroom environment. They require additional speech therapy to enhance verbal skills and should have additional physical activity periods in place of rest periods. These individuals require a structured environment and may need a smaller classroom size for individual attention.
Older children with Prader-Willi syndrome may enter vocational programs (with avoidance of food preparation). Some have attended community colleges.
Patients with Prader-Willi syndrome have a very high threshold for pain and emesis; they can develop ipecac toxicity and have been known to have minimal reports of abdominal discomfort in the presence of significant gastrointestinal pathology such as pancreatitis, acute gastric dilation with necrosis, appendicitis, and cholecystitis.
A goiter
A goiter may present in various ways:Incidentally, as a swelling in the neck discovered by the patient or on routine physical examination
A finding on imaging studies performed for a related or unrelated medical evaluation
Local compression causing dysphagia, dyspnea, stridor, plethora or hoarseness
Pain due to hemorrhage, inflammation, necrosis, or malignant transformation
Signs and symptoms of hyperthyroidism or hypothyroidism
Thyroid cancer with or without metastases
A goiter
Pathophysiology
The thyroid gland is controlled by thyroid-stimulating hormone (TSH; also known as thyrotropin), secreted from the pituitary gland, which in turn is influenced by the thyrotropin-releasing hormone (TRH) from the hypothalamus. TSH permits growth, cellular differentiation, and thyroid hormone production and secretion by the thyroid gland.
Thyrotropin acts on TSH receptors located on the thyroid gland. Serum thyroid hormones levothyroxine and triiodothyronine feed back to the pituitary, regulating TSH production. Interference with this TRH-TSH thyroid hormone axis causes changes in the function and structure of the thyroid gland.
Stimulation of the TSH receptors of the thyroid by TSH, TSH-receptor antibodies, or TSH receptor agonists, such as chorionic gonadotropin, may result in a diffuse goiter. When a small group of thyroid cells, inflammatory cells, or malignant cells metastatic to the thyroid is involved, a thyroid nodule may develop.
A deficiency in thyroid hormone synthesis or intake leads to increased TSH production. Increased TSH causes increased cellularity and hyperplasia of the thyroid gland in an attempt to normalize thyroid hormone levels. If this process is sustained, a goiter is established. Causes of thyroid hormone deficiency include inborn errors of thyroid hormone synthesis, iodine deficiency, and goitrogens.
Goiter may result from a number of TSH receptor agonists. TSH receptor stimulators include TSH receptor antibodies, pituitary resistance to thyroid hormone, adenomas of the hypothalamus or pituitary gland, and tumors producing human chorionic gonadotropin.
Palpation of the goiter is performed either facing the patient or from behind the patient, with the neck relaxed and not hyperextended. Palpation of the goiter rules out a pseudogoiter, which is a prominent thyroid seen in individuals who are thin. Each lobe is palpated for size, consistency, nodules, and tenderness. Cervical lymph nodes are then palpated. The oropharynx is visualized for the presence of lingular thyroid tissue.
The size of each lobe is measured in 2 dimensions using a tape measure. Some examiners make tracings on a sheet of paper, which is placed in the patient's chart. Suitable landmarks are used and documented to ensure consistent measurement of the thyroid gland.
The pyramidal lobe often is enlarged in Graves disease.
A firm rubbery thyroid gland suggests Hashimoto thyroiditis, and a hard thyroid gland suggests malignancy or Riedel struma.
Multiple nodules may suggest a multinodular goiter or Hashimoto thyroiditis.
A solitary hard nodule suggests malignancy, whereas a solitary firm nodule may be a thyroid cyst.
Diffuse thyroid tenderness suggests subacute thyroiditis, and local thyroid tenderness suggests intranodal hemorrhage or necrosis.
Cervical lymph glands are palpated for signs of metastatic thyroid cancer.
Auscultation of a soft bruit over the inferior thyroidal artery may be appreciated in a toxic goiter. Palpation of a toxic goiter may reveal a thrill in the profoundly hyperthyroid patient.
Toxic goiter: A goiter that is associated with hyperthyroidism is described as a toxic goiter.
Examples of toxic goiters include diffuse toxic goiter (Graves disease),
toxic multinodular goiter, and
toxic adenoma (Plummer disease).
Nontoxic goiter: A goiter without hyperthyroidism or hypothyroidism is described as a nontoxic goiter. It may be diffuse or multinodular, but a diffuse goiter often evolves into a nodular goiter. Examination of the thyroid may not reveal small or posterior nodules. Examples of nontoxic goiters include chronic lymphocytic thyroiditis (Hashimoto disease), goiter identified in early Graves disease, endemic goiter, sporadic goiter, congenital goiter, and physiologic goiter that occurs during puberty.
Autonomously functioning nodules may present with inability to palpate the contralateral lobe. Unilobar agenesis may also present like a single thyroid nodule with hyperplasia of the remaining lobe.
The Pemberton maneuver raises a goiter into the thoracic inlet when the patient elevates the arms. This may cause shortness of breath, stridor, or distention of neck veins.
• Iodine deficiency
• Autoimmune thyroiditis - Hashimoto or postpartum thyroiditis• Excess iodine (Wolff-Chaikoff effect) or lithium ingestion, which decrease release of thyroid hormone
• Goitrogens
• Stimulation of TSH receptors by TSH from pituitary tumors, pituitary thyroid hormone resistance, gonadotropins, and/or thyroid-stimulating immunoglobulins
• Inborn errors of metabolism causing defects in biosynthesis of thyroid hormones
Exposure to radiation
Deposition diseases
Thyroid hormone resistance
Subacute thyroiditis (de Quervain thyroiditis)
Silent thyroiditis
Riedel thyroiditis
Infectious agents
Acute suppurative - Bacterial
Chronic - Mycobacteria, fungal, and parasitic
Granulomatous disease
Thyroid malignancy
The different etiologic mechanisms that can cause a goiter include the following:
Correct iodine deficiency and avoid dietary or iatrogenic goitrogens if practical. In the United States, it is difficult to find iodine deficiency, given the supplementation of table salt with iodine, iodine in cattle feed, and the use of iodine as a dough conditioner. Judicious use of levothyroxine is helpful in patients with a previous diagnosis of nodular hyperplasia who have had a lobectomy to prevent occurrences in the contralateral lobe.
Goiter prevention is based on etiology.
Goiters due to autoimmune thyroiditis may be controlled with careful use of levothyroxine and, when indicated, anti-inflammatory medication.
Congenital goiters due to inborn errors of metabolism may be reduced or prevented by careful use of levothyroxine during the postpartum period. Newborns are screened for congenital hypothyroidism.
Complications
Large goiters may cause compression of the trachea, with tracheomalacia and asphyxiation.Hyperthyroidism occurs in some patients exposed to iodine (ie, Jodbasedow phenomenon).
A patient with autoimmune goiters may develop lymphoma. Multinodular goiters may undergo malignant transformation.
Nodular goiters may cause pain, intranodular necrosis, or hemorrhage.
Thyroid abscess may be associated with pain, fever, bacteremia, or sepsis.
• Prognosis
• Benign goiters have a good prognosis. However, all goiters should be monitored by examination and biopsy for possible malignant transformation, which may be signaled by a sudden change in size, pain, or consistency. Fortunately, the risk of this is low. In patients exposed to low levels of radiation the risk rises.• A small percentage of multinodular goiters do cause hyperthyroidism. Lifelong surveillance is necessary.
• Patients with chronic lymphocytic thyroiditis generally have glands that become atrophic
Special Concerns
Withhold treatment of a benign euthyroid goiter in patients who are pregnant until after delivery.Thyroidectomy, if necessary, can be more safely performed in the second trimester.
Small benign euthyroid goiters do not require treatment. The effectiveness of medical treatment using thyroid hormone for benign goiters is controversial. Large and complicated goiters may require medical and surgical treatment. Malignant goiters require medical and surgical treatment.
Treatment
Medical Care
The size of a benign euthyroid goiter may be reduced with levothyroxine suppressive therapy. The patient is monitored to keep serum TSH in a low but detectable range to avoid hyperthyroidism, cardiac arrhythmias, and osteoporosis. The patient has to be compliant with monitoring. Some authorities suggest suppressive treatment for a definite time period instead of indefinite therapy. Patients with Hashimoto thyroiditis respond better.
Treatment of hypothyroidism or hyperthyroidism often reduces the size of a goiter.
Thyroid hormone replacement is often required following surgical and radiation treatment of a goiter. Use of radioactive iodine for the therapy of nontoxic goiter has been disappointing and is controversial.Medical therapy of autonomous nodules with thyroid hormone is not indicated.
Ethanol infusion into benign thyroid nodules has not been approved in the United States, but it is used elsewhere.
Surgical Care
Surgery is reserved for the following situations:• Large goiters with compression
• Malignancy
• When other forms of therapy are not practical or ineffective
Diet
Nutrition plays a role in the development of endemic goiters. Dietary factors include iodine deficiency, goitrogens, protein malnutrition, and energy malnutrition. Often these factors occur concurrently. Iodine: If it is practical, treat endemic goiters in iodine-deficient regions with iodine supplementation in the diet and avoidance of goitrogens. Treatment with iodine supplementation or levothyroxine may reduce goiter size.Goitrogens
Cyanoglucosides are naturally occurring goitrogens that are digested to release cyanide, which is converted to thiocyanate. Thiocyanate inhibits iodide transport in the thyroid and, at higher levels, inhibits organification. Foods that contain cyanoglucosides include cassava, lima beans, maize, bamboo shoots, and sweet potatoes.Thioglucosides are natural goitrogens found in the Cruciferae family of vegetables and weeds eaten by animals. When digested, they release thiocyanate and isothiocyanate, which have thionamidelike properties and are passed to humans via milk ingestion.
Medication
The goals of pharmacotherapy are to reduce morbidity and to prevent complications.
Thyroid hormone replacements
Benign goiters can be treated with thyroid hormone. The most widely used thyroid hormone is levothyroxine sodium, administered once a day. Liothyronine sodium requires more frequent administration. Desiccated thyroid powder, thyroglobulin, and liotrix are less predictable following ingestion.
Levothyroxine sodium (Synthroid, Levoxyl, Levothroid)
Synthetic thyroxine is converted to the active form, triiodothyronine, in the pituitary by 5'-deiodinase. Inhibits production of thyrotropin, which is the main growth factor for the thyroid gland.a multinodular goiter.
The first question is usually: are all the nodules benign? The approach to this question depends on the clinical presentation, associated risk factors, the size of the nodules, and whether the nodules are functioning or non-functioning.Non-functioning or cold nodules within a multinodular gland generally carry the same risk of malignancy as a single isolated cold nodule (10-15% risk of thyroid cancer) and need to be approached diagnostically in a similar manner akin to the investigation of an isolated single cold nodule.
I have a large goitre and borderline high levels of thyroid hormones (mild hyperthyroidism). What are the treatment options?
Traditionally, treatment of patients with large multinodular goitres may include surgery, or radioactive iodine. Not all patients with large goitres have sufficient iodine uptake to allow for effective therapy with radioactive iodine. In some instances, adjuvant use of low dose recombinant TSH may increase iodine uptake in the thyroid and allow for more efficient treatment with radioactive iodine.
Is there any risk to treatment of a multinodular goitre with radioactive iodine?
Patients may experience some degree of neck discomfort and swelling in the region of the thyroid gland following radioactive iodine. Furthermore, destruction of thyroid tissue is often associated with transient worsening of the hyperthyroidism for a few weeks, and in elderly subjects with a history of heart disease, this can be a serious issue. There is some evidence that older subjects treated with radioactive iodine may have an increased risk of mortality, but not if the treatment is sufficiently effective so as to induce hypothyroidism.Clinicians cannot rely exclusively on physical examination to confirm or rule out hypothyroidism. Patients with suspected hypothyroidism require a diagnostic workup that includes thyroid hormone assays.
The combination of signs that had the highest likelihood ratios (coarse skin, bradycardia and delayed ankle reflex) was associated with modest accuracy
physical signs when considered in isolation have poor diagnostic accuracy for hypothyroidism. Even combinations of signs do not appear to have high accuracy. However, since selected signs (such as coarse skin, bradycardia and delayed ankle reflex) are associated with modest accuracy, clinicians could use physical examination to generate and revise their estimates of pre-test probabilities and use the information to select those patients who will benefit most from thyroid hormone assays. This strategy is likely to maximize the number of patients in whom clear diagnostic decisions can be made. The reflex changes corrected with treatment, even before the TSH returned to normal.
long-term Type 1 diabetes may cause delayed muscle contraction and impaired reflex modulation (delayed ankle reflex )which could contribute to gait disturbances and increased number of falls in diabetic patients.
Thyrotoxicosis in pregnancy:
90-95% of cases due to Grave's disease.FETAL RISKS: increased perinatal mortality, 20% are premature, 3% develop overt fetal thyrotoxicosis, 3% biochemical thyrotoxicosis, 3% hypothyroid, 6% minor congenital anomalies.
MATERNAL RISKS: 10% develop cardiac failure.
POST PARTUM THYROIDITIS: occurs in 5% of women. Present with painless goitre and symptoms of thyrotoxicosis 1-3 months post-partum (85% have anti-microsomal antibodies, normal ESR, decresed RAIU). Thyrotoxic phase is very brief (2-5months) and treatment is seldom required. 1/3 become hypothyroid at 4-6 months and may present as postnatal depression. PROGNOSIS: Most are euthyroid at 1 year post-partum. 10-25% recurrence with subsequent pregnancies. Up to 40% will become hypothyroid in the long term (yearly TFT's recommended).
• Improving the Treatment of Necrotizing Pancreatitis
• Acute pancreatitis in most patients is self-limited and resolves without complications or the need for invasive procedures or surgical intervention. In a minority of patients, perhaps 10%, necrosis of the pancreatic and peripancreatic tissues opens the door to multiple organ failure, infection of the necrotic tissue, or both, with a greatly increased risk of death; this risk has been estimated to be 20 to 30
NEJM April 22, 2010
Recent reports citing a substantial reduction in the rate of death, ranging as low as 4%,1,2 attribute the improvement to better intensive care; avoidance of early intervention to allow resuscitation, stabilization, and demarcation and liquefaction of the damaged areas2,3; and a variety of innovations in drainage and evacuation of fluid and devitalized tissues.The established techniques of laparotomy and open débridement1,2,4 are being challenged by "minimally invasive" endoscopic necrosectomy through transgastric, laparoscopic, and retroperitoneal routes.5 The goals in general are control of infection, maximal evacuation of devitalized tissues that are the culture medium for invasive infection, and promotion of conditions for healing.
The indications for surgical intervention certainly do not include the mere presence of asymptomatic necrosis, even if it is substantial.6 Many clinicians argue that infection is the sine qua non,7 but some patients with sterile necrosis nonetheless have a major systemic inflammatory response and multiple organ failure,2,6 and these patients can benefit from débridement. Other patients have a low-grade inflammatory state or they are "persistently unwell" for months,1,2 and as many as 40% of these patients may have unsuspected infection of the necrotic tissue.2
There is now a consensus that the best outcomes from intervention are achieved when débridement is delayed until approximately 4 weeks after the onset of pancreatitis, when the damaged area has been walled off and liquefaction has begun. If necessary, acute sepsis can be preliminarily controlled by percutaneous drainage as a bridge to the later evacuation of detritus. Although percutaneous drainage alone has been associated with some success in the treatment of pancreatic "abscesses," the success rate in definitively treating infected necrotic tissue without the need for débridement is only 30 to 35%.
In this issue of the Journal, van Santvoort et al.9 report the results of a randomized trial comparing treatment of pancreatic and peripancreatic necrosis by open laparotomy with a hybrid, or "step-up," approach in which percutaneous drainage was the first step, with débridement by means of a less invasive video-assisted retroperitoneal débridement (VARD) route reserved for patients in whom the drainage failed.
Participation by 19 institutions was required to accomplish the goals of the trial, and even so, only 88 of 378 patients with necrotizing pancreatitis and confirmed or suspected infection met the inclusion criteria, which included the feasibility of a retroperitoneal access route. A total of 81 of these 88 patients ultimately had proven infected necrotic tissue. Whenever possible, the intervention was not undertaken until approximately 4 weeks after the onset of pancreatitis.
The primary end point selected for the study was a composite of major complications (new-onset multiple organ failure, perforation of a viscus, enterocutaneous fistula, or hemorrhage) or death. With the use of this criterion, the primary end point occurred in only 40% of patients treated with the step-up approach, as compared with 69% of patients who underwent open necrosectomy (P=0.006). However, since the rate of death did not differ between the two groups, the benefits of the step-up strategy accrued from fewer complications (e.g., 5 of 43 patients in the step-up group vs. 19 of 45 patients in the open-necrosectomy group had new-onset organ failure).
Notably, 35% of patients in the step-up group required only the first step (percutaneous drainage), and they apparently tolerated any residual necrotic tissues without a need for débridement. The surprising aspect of this latter observation is that the necrotic tissues left behind must also have been infected, and it is generally assumed that infected necrosis must be removed — in truth, that is the underlying premise for this study.
The authors hypothesize that evacuation of the infected fluid component by means of percutaneous aspiration suffices because the solid debris may be sterile, but it seems more likely that the concomitant antibiotic treatment may succeed in overcoming the residual infection in some patients, as has been anecdotally reported.
Nonetheless, the challenges of adequate débridement were apparent in both groups: one third of each group required multiple attempts (53 attempts in 26 patients who underwent VARD and 91 attempts in 45 patients who underwent open necrosectomy). These figures are significantly higher than the reoperation rate (13%) reported in a recent U.S. study,2 and the length of hospitalization — similar with both methods of treatment — averaged 55 days, which was substantially longer than the 26 to 38 days reported in North American studies.
Part of this longer length of stay may be attributed to differences in clinical practice in the United States and in the Netherlands. However, both techniques included factors that added to the duration of treatment and its costs. Open necrosectomy involved a continuous high-volume lavage through catheters placed at laparotomy until the effluent no longer contained the products of necrosis — a period that could have lasted many weeks.
With the step-up approach, there was frequently an interval of 6 days or longer (median, 10) between the decision to intervene and the first VARD, during which percutaneous aspiration was performed in two trials before failure was declared; all the patients who required VARD also underwent postprocedural lavage. These labor-intensive treatments also added considerably to the overall cost of the treatment and disproportionately biased the reported cost of the group treated by means of open laparotomy in this study.
Despite its limitations, this is an excellent study. The feasibility and probable greater safety of the step-up approach to necrotizing pancreatitis is an important synthesis and integration of evolving techniques. As such, it is a major contribution that is likely to change our practice, at least for a segment of patients with severe acute pancreatitis.
However, its limitations include the narrow applicability of the step-up approach because of the need for a retroperitoneal access route, the need in both groups for multiple operative interventions, and an overall mortality of 17%, which is 50 to 100% higher than the rate of death reported in recent studies from North America.
In addition, as the authors note, this study addresses a comparison of percutaneous drainage plus VARD with open necrosectomy and does not address whether VARD is superior to open necrosectomy when necrosectomy is needed.
The alternative treatment options for necrotizing pancreatitis still require both careful judgment and special skills.
Primary Treatment of Acromegaly with High-dose Lanreotide
• Introduction: The first-line treatment for acromegaly is transsphenoidal surgery. In approximately 50% of patients, however, a cure is not possible with surgery and alternatives are needed. Somatostatin analog therapy is the recommended first-line treatment in patients with such cases. Here we provide the first report of a high-dose lanreotide primary therapy in patients with acromegaly.Case presentation: Six patients who were not suitable for surgery were given 60 mg of lanreotide (Autogel®) every four weeks. All patients were German nationals and Caucasian.
When the response of our patients was unsatisfactory, the dose was increased sequentially to 90 mg every four weeks, 120 mg every four weeks, 120 mg every three weeks and 180 mg every three weeks. Treatment duration was 12 to 24 months. In all cases, the lanreotide dose was 120 mg every 4 weeks or higher. In five of our patients, growth hormone (GH) levels were successfully reduced (in three patients GH <2.5 ng/ml was achieved).
Insulin-like growth factor 1 levels were normalized in three patients and decreased in two patients. One patient failed to show a biochemical response to lanreotide therapy or pegvisomant therapy.Tumor shrinkage or degeneration was observed in the five responding patients. No drug-related adverse events were noted.Conclusions: These results suggest that lanreotide at high doses of 120 mg every four weeks or more is an effective first-line therapy for patients with acromegaly that surgery alone cannot treat.
Introduction
Acromegaly, characterized by elevated growth hormone (GH) and insulin-like growth factor 1 (IGF-1) levels, is associated with a range of cardiovascular, respiratory, endocrine, metabolic and compression symptoms, and with an increased cancer risk.Some symptoms can be serious and life-threatening. If untreated, acromegaly reduces life expectancy.Transsphenoidal surgery is the first-line treatment for acromegaly. However, surgery may be impractical. In approximately 50% of patients, surgery alone is unlikely to control the disease. In cases where there is a low probability of surgical cure, primary treatment with a long-acting somatostatin analog is recommended.[6,7]
Two somatostatin analogs are available, such as octreotide (Sandostatin®, Novartis) and lanreotide (Autogel®, Ipsen). There are considerable clinical data on the first-line use of octreotide (for example, Colao et al. [8]). Lanreotide, on the other hand, has been shown to be effective as secondary treatment at starting doses of 30 mg to 120 mg every four weeks,[9] but only recently have data been published on the use of lanreotide 90 mg or 120 mg every four weeks as primary therapy.
We present six patients with acromegaly who received primary treatment with lanreotide at doses higher than those presented in the literature.
GH, IGF-1 and prolactin levels were measured using chemiluminescent immunometric assays with Immulite 2000 (Siemens Medical Solutions Diagnostics, formerly DPC, Los Angeles, USA). Normal GH levels were defined as 0.5 to 5.0 ng/ml and normal age-adjusted IGF-1 levels were between 81 and 483 ng/ml.
Magnetic resonance tomography was conducted with a 1.5 Tesla-System (Siemens, Erlangen) Type AVANTO, and 1.5 T. MR (Avanto, Siemens). Magnetic resonance imaging (MRI) images were produced using a head matrix array. For pituitary imaging, T2w-TSE sag, T1w-SE sag and coronar before and after contrast medium gadolinium (Gadovist, Bayer Healthcare) were used dynamically. The slice size was 2.2 mm.
The six patients with acromegaly presented in this case series were ineligible for surgery because three of them had macroadenoma that were too large and had parasellar and suprasellar extensions (Patients 1, 5 and 6), while one had an adenoma that was too close to the internal carotid artery (Patient 2). One had cardiac insufficiency stage New York Heart Association (NYHA) III and severe insulin-dependent diabetes mellitus and was thus considered high risk for systemic anesthesia (Patient 3), while one had McCune-Albright syndrome and fibrous dysplasia involving the base of the skull which made transsphenoidal surgery impossible (Patient 4).
All patients were German nationals and Caucasian.All patients initially received lanreotide by deep subcutaneous injection (Autogel®, Ipsen, Paris, France). Each patient was given a single starting dose of 60 mg. After four weeks, their IGF-1 and GH levels were measured. If the IGF-1 levels remained high and the response of our patient to the medication was unsatisfactory, the next dose of lanreotide was increased to 90 mg or 120 mg.
If a patient who was receiving treatment with 120 mg, had an unsatisfactory IGF-1 response, the injection interval was reduced to every three weeks. If GH and IGF-1 levels were still elevated in patients receiving lanreotide 120 mg every three weeks, the dose was increased to 180 mg, or 90 mg in each gluteal muscle, every three weeks. For each patient, the dose adjustments were usually made after three injections of lanreotide. In single cases with very high IGF-1-levels, the decision to increase the dose was made earlier based on our previous experience with this treatment
• Conclusions
• This series of six patients with acromegaly is one of the first specific reports of primary treatment with lanreotide, and the first to report the use of high doses. The six patients were not eligible for surgical removal of their pituitary tumors, and five patients showed a biochemical response to lanreotide treatment, which was given for one to two years.• The response of our patients to lanreotide treatment occurred within one to two months, but required dose increases to 120 mg every four weeks, while two patients
subsequently required 180 mg every three weeks to achieve or maintain their initial response. Furthermore, the five patients that responded also had evidence of tumor shrinkage or degeneration while receiving lanreotide.
The goals of treatment for acromegaly are to reduce GH levels to <2.5 ng/ml, normalize IGF-1 levels, and/or control tumor mass.[6,7] Based on these goals, lanreotide was proven to be a successful first-line therapy in five of these six patients, with the exception of Case 3, as this patient did not respond to high-dose lanreotide or to pegvisomant.
The other unique aspect of this case series is the high dose of lanreotide (180 mg every three weeks) that was given to two of our patients. This high dose of lanreotide has now been given to one patient for approximately six months and to another for approximately three months with no unexpected adverse events.
The use of such high doses of lanreotide has not been previously published, and our experience suggests that this dose is well-tolerated, at least in the short-term, and may be useful for patients showing an attenuation of response to lanreotide doses of 120 mg every four weeks.
This report confirms the efficacy and tolerability of lanreotide in the primary treatment of acromegaly. Further data are required regarding the use of lanreotide in this setting, as well as to identify the potential clinical risks and benefits of high-dose lanreotide.
Men and women should be routinely asked about their sex life, say new guidelines on the management of testosterone deficiency and sexual disorders.
Doctors behind the guidelines say that patients and doctors lack awareness of and are reluctant to discuss sexual problems and that the importance of sexual function for general health needs to be more widely recognised.
BMJ 2010;341
because tests are not sensitive enough to detect the low concentrations in women and the normal range in women has not been established. However, assessing testosterone deficiency in men should be made over several consultations, say the guidelines. The diagnosis should be based on symptoms such as poor morning erection, low sexual desire, and erectile dysfunction, combined with a measure of testosterone concentrations in blood taken in the morning.
Testosterone testing in women is not recommended
Geoff Hackett, one of the coauthors and a consultant in sexual medicine at the Good Hope Hospital in Birmingham, said that low testosterone contributes to erectile dysfunction in about 20% of men and that it is the sole medical cause of sexual problems in 10%.
Half the men who are getting repeat prescription of sildenafil from their GP are failing to achieve adequate sexual function, Dr Hackett told a press briefing on 24 September, and for many the reason is low testosterone concentrations.
When men find out that their problem is low testosterone they often feel angry that they have spent a lot of money on sildenafil, or a drug like it, and that their hormone concentrations had not been measured earlier, he said.
The guidelines say that all women with problems of sexual desire or arousal should be offered psychosexual counselling or sex therapy and that oestrogen, testosterone, or tibolone (a synthetic steroid often used in postmenopausal women) should be offered, as appropriate.
Kevan Wylie, consultant in sexual medicine at the Royal Hallamshire Hospital in Sheffield and lead author of the guidelines, said, “Testosterone is an important hormone to the body in that it affects not only sexual function but also general wellbeing. All treatments for sexual disorders should be individually tailored, depending on case history and clinical symptoms. The need for testosterone replacement therapy in men and women needs to be carefully assessed.”
The signaling mechanisms used by sperm — to capacitate, migrate to the fertilization site, penetrate the thick layers of cumulus oophorus and the zona pellucida, fuse with the egg membrane, and activate the egg — are poorly understood at the molecular level. A recent study by Lishko and colleagues1 describes the measurement of electrical signals in human sperm and a signaling pathway pertinent to male fertility.
Sperm and the Proton Channel
The detailed molecular workings of hundreds of ion channels have been extensively studied in somatic cells over past decades, and mutations in genes encoding these channels have been linked to numerous genetic diseases. Progress in ion-channel research in sperm, however, has been frustratingly slow.
A major problem has been that whole-cell patch-clamp recording, a technique used for decades to monitor the activity of ion channels in somatic cells, has only recently been successfully applied to sperm (in the mouse). Subsequently, Lishko and colleagues used this technique to measure the electrical current of human sperm. By forming a tight seal between a glass pipette and a small patch of sperm membrane and breaking the isolated membrane, they could monitor the electrical activity of the sperm cell.
A major purpose of ion channels is to regulate the intracellular Ca2+ concentration. Sperm use Ca2+ to control fundamental processes ranging from capacitation to the acrosome reaction and the penetration of the egg coat. Unique to sperm are a family of alkalinization-activated Ca2+-permeable ion channels called the CATSPER channels.2 Ca2+ influx through CATSPER channels is required for hyperactivated motility of sperm, without which sperm can neither migrate to the fertilization site nor penetrate the egg coat to achieve fertilization.
An alkalinization-activated K+ channel (KSPER), formed by the slowpoke homolog 3 (SLO3) protein (also called KCNU1), is also specific to sperm. Both types of channels are localized in the principal piece of sperm tail and each is specifically required for male fertility.
In the excitable cells of nerve and muscle, changes in membrane voltage during action potentials cause voltage-gated channels to open and close. Sperm do not have action potentials, however. Rather, changes in intracellular pH and in concentrations of other key messengers result in the opening and closing of channels.
During the sperm's journey in the female reproductive tract, the environmental pH changes dramatically, from approximately 4 to approximately 7, corresponding to a reduction of the extracellular H+ concentration by a factor of approximately 1000. Moreover, sperm maintain a low pH when quiescent in the epididymis; in fact, a low pH helps to maintain their quiescence. They undergo intracellular alkalinization during capacitation and when stimulated by physiologic stimuli such as the egg coat.
How do sperm cells become alkalinized?3 One potential mechanism is through Na+–H+ exchangers that pump H+ out in exchange for Na+. In particular, the sperm-specific Na+–H+ exchanger, which is localized in the sperm tail along with the CATSPER and KSPER channels and is required for male fertility, may be responsible for alkalinization.4
However, the discovery by Lishko and colleagues moves the spotlight onto the proton channel HV1, which is involved in the extrusion of H+ and therefore alkalinization (Figure 1). HV1 channels are controlled by changes in the transmembrane voltage and pH gradients, and the channels selectively conduct H+.
The physiologic function of these channels has been characterized best in phagocytic leukocytes, in which they provide a pathway to H+ efflux and to charge compensation for NADPH oxidase during the respiratory burst of superoxide production.5 Unlike transporters, the movement of H+ through the HV1 channel is passive, so the use of such a channel for intracellular alkalinization is limited to times when the membrane potential is positive with respect to the equilibrium potential for protons. In contrast to human sperm, mouse sperm have a minimal Hv1 current, and mice lacking the Hv1 gene are fertile.5
In response to alkalinization, KSPER channels open to allow for K+ efflux and hyperpolarization of the cell, while CATSPER channels open to allow for Ca2+ entry to trigger hyperactivated motility. A Na+–H+ exchanger pumps H+ out to maintain alkalinization. CATSPER, KSPER, and the Na+–H+ exchanger are all sperm-specific, and disabling mutations in their corresponding genes result in complete infertility in male mice; mutations in CATSPER proteins result in complete infertility in men.
Etiology
Corticotropin-releasing hormone (CRH), produced by the hypothalamus, stimulates the production and release of corticotropin (ACTH) through its actions on the anterior pituitary. ACTH then acts on the adrenal cortex to release cortisol (FIGURE 1).[7]Cushing's Syndrome
Cortisol regulates the breakdown of fat and the metabolism of proteins and carbohydrates. Cortisol production increases prior to waking and peaks within a few hours after waking. Cortisol levels are lowest before sleep and during the first few hours of sleep.
Hypercorticism refers to an excess of corticosteroids in the body. This excess of corticosteroids—specifically, cortisol—can lead to CS. The excess cortisol is rarely due to spontaneous cortisol production by the adrenal gland. Typical causes include tumors and drugs.
A patient who has CS may present with one or more of the following: moon face, central obesity (trunk accumulation of adipose tissue), myopathy, muscle weakness, fatigue, hirsutism, osteoporosis, amenorrhea, hypertension, psychiatric symptoms, striae (purple stripes or lines in skin), back pain, and/or diabetes mellitus. CS can have ACTH-dependent or ACTH-independent origins.
ACTH-Dependent CS
Excessive cortisol release due to increased ACTH levels is defined as ACTH-dependent CS. A major cause of ACTH-dependent CS is the presence of ACTH-dependent tumors normally found in the pituitary gland. Pituitary adenomas, which account for approximately 70% of dependent causes, are typically small and benign. Ectopic tumors are the other major cause of ACTH-dependent CS. These ectopic tumors occur in abnormal places (i.e., lung, kidney) and account for about 15% of ACTH-dependent CS cases.Ectopic tumors outside the hypothalamus constitute less than 1% of ACTH-dependent cases. These nonhypothalamic tumors excrete CRH, which leads to the overproduction of ACTH.
ACTH-Independent CS
Cases of ACTH-independent CS present with increased cortisol levels that are not related to increased ACTH levels. ACTH-independent CS is further broken down by cause—either exogenous or endogenous.Exogenous CS: Drug-induced, or exogenous, CS occurs when an excess of steroids is introduced into the body. Drug-induced CS is the most common cause of the syndrome.
Endogenous CS: There are several types of ACTH-independent tumors. Adrenal adenomas and carcinomas are the most common independent tumors. Other independent causes, occurring in less than 1% of cases reported, include bilateral macronodular hyperplasia and primary pigmented nodular adrenocortical disease
Diagnosis
To diagnose CS, a history is obtained and a physical examination is conducted to assess the presenting signs and symptoms. There are currently two first-line screening tests for CS, with a third screening test currently being validated. The first-line screening tools are the 24-hour urinary free cortisol (UFC) test and the low-dose dexamethasone suppression test (DST).Confirmation of CS
UFC Test: UFC levels correlate with free plasma levels of cortisol. The patient collects three 24-hour samples of urine for analysis.Normal urine cortisol tests are under 100 mcg/24 h.A result that is four times the upper limit of normal (typically 440 mcg/24 h), which is rare, indicates CS.TheUFC test is not indicated in patients with renal impairment—defined as a glomerular filtration rate less than 30 mL/min—because of decreased cortisol excretion. The screening sensitivity does not allow for the detection of subclinical or preclinical CS.
DST: In healthy patients, dexamethasone suppresses plasma cortisol levels. In CS, this mechanism can be overridden by the underlying disease. For the DST, the patient is administered 1 mg dexamethasone between 2300 and 2400 hours. The following morning, cortisol concentration levels are measured. A measurement above 5 mcg/dL indicates inhibition of a negative feedback loop, as seen in CS. While a level of 1.8 mcg/dL allows for identification of pseudo-CS, cyclic CS, or early stages of CS, it also may create false-positive results. A two-day 2-mg DST may be used in clinical practice; however, cost and increased hospitalization are limiting factors in its use.
Late-Night Salivary Cortisol Test (LST): LST is a new screening tool being evaluated for CS. Salivary cortisol concentration is closely related to free plasma, but it remains independent of salivary flow rate. Normal ranges for this screening are assay-dependent. Current studies suggest high specificity and sensitivity, but further research is needed to validate the use of LST as a screening tool.[10]
Differential Diagnosis
Once CS is diagnosed, further testing is necessary to differentiate the cause, thereby enabling proper treatment. Current second-line tests are ACTH measurement, high-dose DST, and the CRH stimulation test (TABLE 1).[10] All patients with ACTH-dependent CS should undergo pituitary MRI.The bilateral inferior petrosal sinus sampling (BIPSS) test samples blood surrounding the pituitary, allowing for accurate diagnosis of pituitary origin of high ACTH. BIPSS also is recommended in patients with ACTH-dependent CS. However, owing to rare but significant complications, the risks and benefits need to be taken into consideration. BIPSS has a sensitivity of 95% to 99% when performed at "experienced centers."Several new differential diagnosis tests are currently being researched, including one involving the use of desmopressin.
Complications of CS
Accurate treatment of CS is important in order to avoid the numerous complications that can stem from disease progression. Major cardiovascular complications are hypertension, impaired glucose tolerance, obesity, hyperlipidemia, hypercoagulability, metabolic syndrome, and osteoporosis.Psychiatric complications such as psychosis, mania, suicidal tendency, obsessive-compulsive disorder, cognition changes, and anxiety also may develop. Last, high cortisol levels may create alterations in other areas of the endocrine system, such as the somatotrophic axis (which can decrease growth rate in younger patients or alter fat distribution in adults), the gonadal axis (which can have adverse effects on reproductive organs), and the thyroid axis (which can lead to hypothyroidism or hyperthyroidism). These complications may persist even if a patient has subclinical CS.
Treatment
Drug-induced CS typically is treated by discontinuing the causative medication. Problems arise in complex patients (e.g., transplant patients), in whom discontinuation of the steroid medication may lead to serious adverse outcomes. Surgery, radiation, and pharmacotherapy constitute the standard treatment for tumor-related causes of CS. Following treatment, further screenings are necessary to assess the success of therapy.Management of Cushing's Syndrome:
Nonpharmacologic
The first-line option for pituitary tumors is transsphenoidal surgery (technique in which the pituitary tumor is removed through the nose, leaving remaining pituitary function intact), which gives the patient a chance of remission of CS. Surgical remission rates range from 60% to 80%, but this number may vary owing to different definitions of remission. The average recurrence rate is around 20%. As with surgical remission, there is no current collaborated definition of a cure for CS. Surgery or radiation with or without adjunctive pharmacologic treatment is currently seen in clinical practice.Pharmacologic
Current pharmacologic treatment does not correct the underlying cause of excessive cortisol, which typically limits its use to adjunctive therapy. However, pharmacologic monotherapy may be necessary when surgery or radiation is not appropriate or is delayed. Three pharmacologic strategies exist to treat CS.
Inhibition of Steroid Synthesis: The first strategy is the inhibition of steroid synthesis. Drugs used in this strategy include ketoconazole, mitotane, metyrapone, and etomidate.
These drugs, through different mechanisms, block the conversion of cholesterol to cortisol. This causes a decrease in cortisol levels and the resolution of CS symptoms.
There are several disadvantages to this strategy: Higher-than-normal doses of medications are used, patients rarely remain in remission after discontinuing the medication, and steroid replacement therapy may be needed to avoid adrenal insufficiency. None of these agents is FDA-approved to treat CS, and the doses reported in the literature cover a wider-than-normal range, indicating the need for very careful attention to side effects.
Ketoconazole. A common side effect of ketoconazole, an imidazole antifungal agent, is inhibition of the CYP450 pathway. This inhibition halts the production of cortisol, leading to a reversal of CS symptoms. Ketoconazole therapy is initiated at a daily dose of 400 mg to 600 mg. The dose is titrated until a level of 1,200 mg to 1,600 mg, divided into four equal doses, is reached.
Ketoconazole's safety and efficacy support its use as a first-line choice for many prescribers.
Mitotane. An antineoplastic agent, mitotane (Lysodren) inhibits cortisol synthesis by inhibiting 18-hydroxylase and 3-beta-hydroxysteroid dehydrogenase. At high doses, mitotane has adrenolytic activity (breaks down adrenaline), which causes adrenal cortical atrophy. Mitotane is started at 0.5 g to 1 g daily and increased every 1 to 4 weeks.[13] At doses greater than 4 g daily, the adrenal cortex may be irreversibly impaired. Mitotane's half-life is 18 to 159 days, and the drug has been detected in adipose tissue as long as 22 months after treatment.
]
Metyrapone. Metyrapone (Metopirone), a diagnostic agent, works by blocking 11-beta-hydroxylase, halting cortisol production. Metyrapone can be used as monotherapy or as adjunctive therapy with radiation. Typical dosing of metyrapone begins at 0.5 g to 1 g daily, increasing every few days to a maximum dose of 6 g.[13] Blocking 11-beta-hydroxylase leads to increased ACTH, resulting in increased androgenic and mineralocorticosteroid precursors. This can lead to adverse effects such as hypertension, acne, hirsutism, and nausea. Although this agent is not yet available in the U.S., compassionate use is an option.
Etomidate. Etomidate (Amidate), an imidazole-derivative anesthetic, is used for the induction and maintenance of anesthesia. It blocks cholesterol's side-chain cleavage and 11-beta-hydroxylase, inhibiting the conversion of cholesterol to cortisol. Etomidate is the only medication in this strategy that can be given IV, and case reports have cited a significant reduction in cortisol within 12 hours. Because of its anesthetic effects, careful dose titration is required. An effective dosage range has been reported to be 0.01 mg/kg/h to 0.1 mg/kg/h.[18]
Modulation of Pituitary ACTH: The second strategy is to modulate pituitary ACTH release, causing a decrease in cortisol secretion from the adrenal gland. This strategy has a poor response rate, and no large-scale, placebo-controlled studies have been reported. As with the drugs used to inhibit steroid synthesis, none of these agents is FDA-approved for the treatment of CS. Drugs used include cyproheptadine, octreotide, and valproic acid.
Inhibition of Cortisol's Action: The third strategy involves using an agent that blocks cortisol's action. Mifepristone competitively inhibits glucocorticoid, androgen, and progestin receptor binding. No large clinical trials have been performed to assess the benefit of this drug, but a few investigational studies of ectopic CS have shown a potential benefit. However, dosage adjustment was found to be difficult. Currently, mifepristone is indicated only as an abortifacient in the U.S. Its use in CS is an off-label indication, and further testing is warranted.

nejm.org 550 february 10, 2011
Hyperthyroidism is predominantly a disorder in women. Overt hyperthyroidism, definedas subnormal serum thyrotropin concentrations with elevated free T3 or free T4 concentrations, has a prevalence of approximately 0.6% among women. In areview of several large studies, the incidence of hyperthyroidism was approximately
0.4 cases per 1000 women per year; the incidence in men was 25% or less of the incidence in women.
Subclinical hyperthyroidism, defined as subnormal levels of serum thyrotropin with normal levels of free T3 and free T4, has a prevalence
of 0.7% in the United States. Graves’ disease is the most common cause of hyperthyroidism
in all age groups in the United States, but for areas in which the population has iodine deficiency, the prevalence of toxic adenoma and multinodular goiter
increases with age, and these disorders are more common than Graves’ disease in older persons.
Graves’ disease is an autoimmune disorder
caused by an antibody that acts as an agonist onthe thyrotropin receptor. The result is unregulated
synthesis of thyroid hormone. Spontaneous
remission occurs in approximately 30% of patients.
Ophthalmopathy characterized by inflammation
of periorbital and retro-orbital connective
tissue, fat, and muscle is clinically
evident in approximately one third of patients and
is unique to Graves’ disease. Thyrotropin receptors
are found on orbital fibroblasts and adipocytes;
they are probably the autoimmune target
responsible for Graves’ ophthalmopathy.
Toxic adenoma and multinodular goiter are not associated with ophthalmopathy;
these conditions do not resolve spontaneously unless the hyperthyroidism was precipitated by administration of pharmacologic amounts of iodine or, in rare cases, by the spontaneous infarction of a toxic adenoma.
In normal thyroid tissue, thyrotropin is required to stimulate iodine uptake into follicular cells.
Since thyrotropin-receptor antibodies activate the thyrotropin receptors in Graves’ disease,radioiodine is concentrated within the entire gland.
In contrast, in toxic adenoma or toxic nodular goiter, thyrotropin is suppressed by the hyperthyroid state, and radioiodine is concentrated only in tissue that is autonomous (i.e., not subject to normal regulation by thyrotropin) (Fig. 1B).
However, if the patient is treated with antithyroid
drugs before radioiodine administration and if thyrotropin levels are allowed to become normal or high, then radioiodine will be concentrated in both the autonomous and normal thyroid tissue.
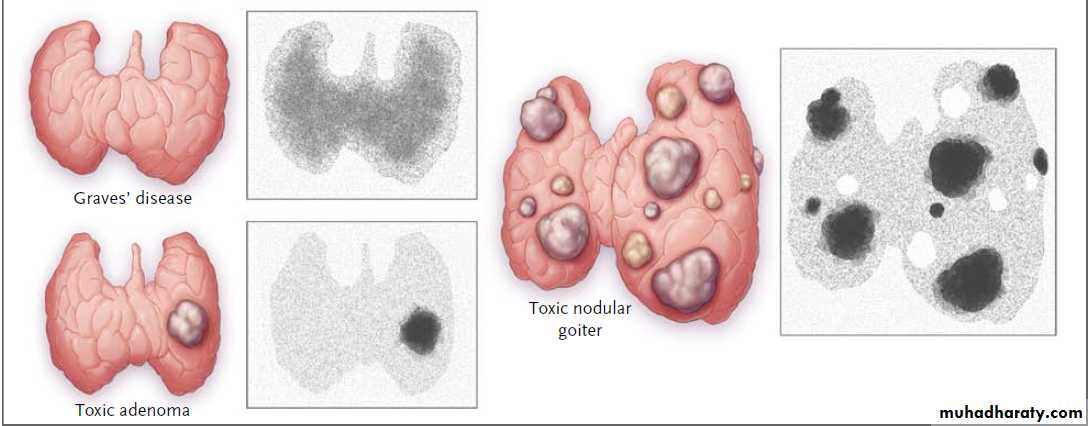
Three efficacious treatments are available for hyperthyroidism.
Radioiodine therapy and surgeryare considered definitive therapies, since the primary goal of this approach to treatment is to
destroy hyperfunctioning thyroid tissue. Antithyroid drugs (methimazole, carbimazole, and propylthiouracil) are used in one of two ways.
On occasion before radioiodine administration
And usually before surgery, several weeks of treatment with an antithyroid drug is administered to achieve a euthyroid state. Antithyroid drugs are
also used in Graves’ hyperthyroidism for 1 to 2
years, or longer, with the hope of achieving a remission.
Remission of hyperthyroidism is not
expected when antithyroid drugs are used during
the long-term treatment of toxic adenoma or toxic
multinodular goiter;
in these cases, early definitive
therapy with radioiodine or surgery is indicated.
Absolute contraindications to radioiodine treatment
are pregnancy, lactation, and an inability
to comply with radiation safety regulations. Radioiodine is considered safe for use in women of
childbearing age and in older children.
Patients who are allergic to iodinated radiocontrast agents are usually not allergic to radioiodine. Moderately severe ophthalmopathy may be
a contraindication to treatment with radioiodine,
since radioiodine may exacerbate the condition.
Radioiodine Therapy for Hyperthyroidism
nejm.org 550 february 10, 2011Worsening ophthalmopathy is more common
in cigarette smokers than in nonsmokers.Concurrent administration of glucocorticoids
mitigates exacerbations, at least in patients
with mild ophthalmopathy. ophthalmopathy worsened in 10% of the patients receiving methimazole, 16% of those undergoing surgery, and 33% of those treated with radioiodine.
Beta-adrenergic blocking agents, if not contraindicated,are usually administered in order to ameliorate symptoms.
Most endocrinologists also treat highrisk
patients with antithyroid drugs for several weeks before administering radioiodine in an attempt to achieve normal or near-normal thyroid function as rapidly as possible. High-risk patients include elderly persons, patients with underlying cardiovascular disease, and patients with severe hyperthyroid symptoms or with concentrations of thyroid hormone that are two to three times as high as the upper limit of the normal range.
Pretreatment with an antithyroid drug may
increase the risk of treatment failure with the initial
radioiodine dose; in one study, this occurred
after the administration of propylthiouracil but
not methimazole. The use of higher doses of
radioiodine can compensate for this effect.
Antithyroid drugs are discontinued 2 to 3 days
before the administration of radioiodine.
Radioiodine is given orally as a single dose of
131I-labeled sodium iodide (Na131I) in liquid orcapsule form. One randomized trial suggested the
superiority of calculated-dose regimens, which
take into account the estimated thyroid weight
in grams, the desired dose (100 to 200 μCi per
gram), and the 24-hour radioiodine uptake. Another
study, however, reported that the use of
three fixed doses in amounts based on gland size
as determined by palpation (5, 10, or 15 mCi [185,
370, or 555 MBq]) was as effective.
I generally prefer the use of the calculated-dose approach,since the radioiodine uptake test can also be used to help determine the cause of the hyperthyroidism.Iodine is rapidly absorbed, is concentrated in the thyroid, and is cleared by the kidneys or through hemodialysis. Clearance of radioiodine is that reduced in patients with renal failure, which meansbody organs and bone marrow are effectively exposed to higher doses.
The cell necrosis induced by radioiodine occurs
gradually, and an interval of 6 to 18 weeks or longer must elapse before a hypothyroid or euthyroid state is achieved. During that interval,hyperthyroidism may transiently worsen before it resolves.If the patient was pretreated with antithyroid
drugs, they may be resumed 3 to 7 daysafter radioiodine administration, and treatment
with beta-adrenergic blocking agents is typically
continued. The patient’s thyroid function is monitored at intervals of 4 to 6 weeks. When thyroid function has normalized, treatment with betablockers and antithyroid drugs is stopped and
levothyroxine is administered as indicated, with
the goal of maintaining normal thyroid function.
Suppression of serum thyrotropin may be prolonged after successful treatment; therefore measurement of free T4 and T3 is essential for several months after radioiodine therapy.
If sufficient radioiodine is administered, hypothyroidism develops in 80 to 90% of patients with Graves’ disease; 14% of patients require additional treatment.
Failure of the first dose to cure hyperthyroidism
and the need for a second courseof treatment may be obvious after an interval as
short as 3 months, if the gland size fails to regress
and thyroid hormone levels remain quite
high. However, if the degree of hyperthyroidism
is minimal 3 months after the initial treatment,
hormone levels may slowly approach the normal
or hypothyroid range, obviating the need for retreatment.
Attainment of normal thyroid function after
asingle dose of radioiodine is an elusive goal.
Continued stimulation of thyrotropin receptors in
nonablated tissue may cause recurrent hyperthyroidism.
More commonly, hypothyroidism occurs
well after treatment has concluded. In patients with Graves’ disease, complete ablation of the
gland is the goal of radioiodine treatment.
In patients with toxic adenoma, the goal of
therapy is to ablate only the adenoma.
If radioiodine is administered when the thyrotropin
level is suppressed as a result of the patient’shyperthyroid state, it does not become concentrated
in normal extranodular tissue and the
patient should be euthyroid after ablation of the
adenoma. However, hypothyroidism is likely if
the patient is pretreated with antithyroid drugs
and thyrotropin levels are not suppressed at the
time of treatment, allowing radioiodine to become
concentrated in normal thyroid epithelium.
In patients with toxic nodular goiter, the outcome depends on the extent of autonomous
thyroid tissue and the thyrotropin level at the
time of treatment. However, another goal may be
to reduce goiter size: normal or elevated levels of
thyrotropin facilitate iodine uptake into both
autonomous and nonautonomous thyroid tissue,
reducing goiter size but increasing the risk of
hypothyroidism.
Retreatment with radioiodine is necessary in
10 to 30% of patients with toxic adenoma andin 6 to 18% of patients with toxic nodular goiter.
Hypothyroidism occurs in only 3% of patients
after 1 year of treatment but is present in
24 to 60% of patients after 20 to 24 years, and it
occurs more commonly in patients who also have
Hashimoto’s thyroiditis. Radioiodine reduces
goiter size by 40%37 and improves compressive
symptoms in slightly less than 50% of patients.
In the United States, radioiodine is administered
on an outpatient basis, but some countries,depending on the dose, require several days of
hospitalization because of radiation safety concerns.
Currently, radiation safety requirements
vary from state to state and more markedly from
country to country, and they may depend on the
administered or retained radiation dose.
Typically,patients are advised to avoid contact with children and pregnant women for up to a week or longer, to restrict close contact with nonpregnant adults for several days (e.g., a 2-hour limit for contact at 1.8 m [6 ft] or closer), and to be meticulous about toilet use and cleaning .
Adverse Effects
Radiation thyroiditis, a painful inflammation ofthe thyroid, occurs in 1% of patients and lasts a
few weeks. This condition is frequently associated
with exacerbation of thyrotoxicosis because of
the destruction-mediated release of thyroid hormone.
Treatment typically consists of nonsteroidal
antiinflammatory drugs and beta-adrenergic
blockers, but some patients require glucocorticoids
to relieve the pain.
In up to 5% of patients with toxic nodular goiter, Graves’ disease develops after radioiodine therapy. It is likely that the release of thyroid antigens from inflamed tissue triggers the production of thyrotropin-receptor antibody.
This condition is treated with a second dose of radioiodine.
In most studies, radioiodine has not been associated with an increased risk of cancer.
The Cooperative Thyrotoxicosis Therapy Follow-up Study Group has followed 35,593 patients after treatment for hyperthyroidism, and after a mean follow-up of 21 years, the group reported no increase in the overall risk of death from cancer among patients who received radioiodine. There was a small increase in the risk of thyroid cancer, primarily among patients with toxic nodular goiter,
which was thought to be partly due to the underlying disease rather than the treatment.
In another study, which for 36 years followed 107
patients with hyperthyroidism who had beentreated before the age of 20 years, no increased
risk of cancer was reported. A British study of
7417 patients reported a reduced risk of cancer
overall but a slight increase in the risk of thyroid
and colon cancer. A Finnish study of 2793 patients
reported an increased risk of stomach, kidney,
and breast cancer.
Patients receiving radioiodine therapy are at
increased risk for death from cardiovascular disease
primarily in the first year after treatment.
This is thought to be a consequence of the thyrotoxic
state rather than the treatment.
Nodular thyroid tissue may regress after radio iodine ablation, but it rarely disappears, and over
time calcifications may develop. Calcified nodules
are evident on subsequent imaging and may
be viewed as possibly precancerous by radiologists,
which may lead to thyroid surgery
Areas of Uncertainty
There is considerable uncertainty about the optimaltherapy for hyperthyroidism, owing to the
absence of data from large, well-designed, randomized,controlled trials. Moreover, the perception of what constitutes optimal therapy appears to differ dramatically among physicians, patients,and professional societies, being highly dependent on the relative values placed on risks and outcomes.
Questions that could be addressed by welldesigned
studies include the following:Which patients receiving radioiodine require pretreatment with antithyroid drugs?
When patients are first treated with antithyroid drugs, should the drugs be reinstated after treatment with radioiodine,and if so, at what interval?
What are the optimal subsequent interventions, and what are the costs of follow-up after radioiodine therapy for patients with toxic nodular goiter?
Controversy also surrounds guidelines for radiation safety after radioiodine therapy.
For example,in 2008, the Nuclear Regulatory Commission released a statement regarding the protection of children who may come into contact with patients treated with radioiodine. In this statement,it was suggested that physicians should
consider keeping patients hospitalized if they live
with infants or young children.
Some experts argued that this recommendation, although not a mandate, would result in unnecessarily restrictive management of patient care. Uncertainty about this and many other issues related to the safe and appropriate use of radioiodine persist because there are few direct data on which to base firm recommendations.
Guidelines from Professional Societies
The draft version of the guidelines on the management of hyperthyroidism from the American Thyroid Association and the American Association of Clinical Endocrinologists, expected to be published in final form in 2011, states that radioiodine, antithyroid drugs, and surgery are all reasonable treatment options for hyperthyroidism (Bahn RS: personal communication). These guidelines emphasize the importance of detailed discussions with the patient regarding each option’s
benefits and risks relative to coexisting conditions,
lifestyle, and the patient’s values.
Pretreatment with antithyroid drugs should be considered in elderly persons and in patients with underlying cardiovascular disease, severe hyperthyroid symptoms, or thyroid hormone concentra-tions that are two to three times the upper limit of the normal range.
Surgery, rather than radioiodine
therapy, is recommended for patients with
active, moderately severe Graves’ ophthalmopathy.
Concurrent use of glucocorticoids should be considered in those with active, mild ophthalmopathy
and in smokers.
Children are usually treated with methimazole for 1 to 2 years, but treatment with radioiodine, surgery, or methimazole is appropriate
for children over 10 years of age.
Pregnancy is typically postponed for 6 months after radioiodine treatment.
The patient might choose surgery either to
shorten this delay or because of concern that exposure to radioiodine could exacerbate the ophthalmopathy.

Don't drink your calories
Health tip
