
Diagnosis and management of thalassaemia
BMJ 25 January 2012د. حسين محمد جمعه
اختصاصي الامراض الباطنة
البورد العربي
كلية طب الموصل
2012
Increasing global migration has resulted in wider dispersal of people at risk of hereditary anaemias. As a result, haemoglobinopathies are becoming increasingly prevalent in countries where these diseases are not endemic. The treatment
of thalassaemia major and intermedia has traditionally depended on preventing undesirable outcomes of disease, using transfusion therapy along with iron chelation.
The only cure for the disease is stem cell transplantation. However, this is a complicated procedure, with better outcomes when offered at young ages,
which strengthens the desirability of screening newborns in high risk populations.
Multidisciplinary management of thalassaemia is recommended by international guidelines.
Antenatal prevention programmes consist of identifying and counselling couples who carry the relevant genes, and offering them different options to prevent having a child with thalassaemia. In this review we summarise diagnostic, treatment, and prevention options in thalassaemia for generalist readers.
What is thalassaemia?
The thalassaemias are a group of recessively autosomal inherited conditions characterised by decreased or absence of synthesis of one of the two polypeptide chains (α or β) that form the normal adult human haemoglobin molecule (haemoglobin A,α2/β2), which results in reduced haemoglobin in red cells and anaemia.The term thalassaemia derives from the Greek words
thalassa (sea) and haima (blood) and the thalassaemiasyndromes are named according to the globin chain affected or the abnormal haemoglobin involved; β globin gene defects may give rise to β thalassaemia, while mutations of the α globin gene may cause α thalassaemia.
Classification is by clinical severity
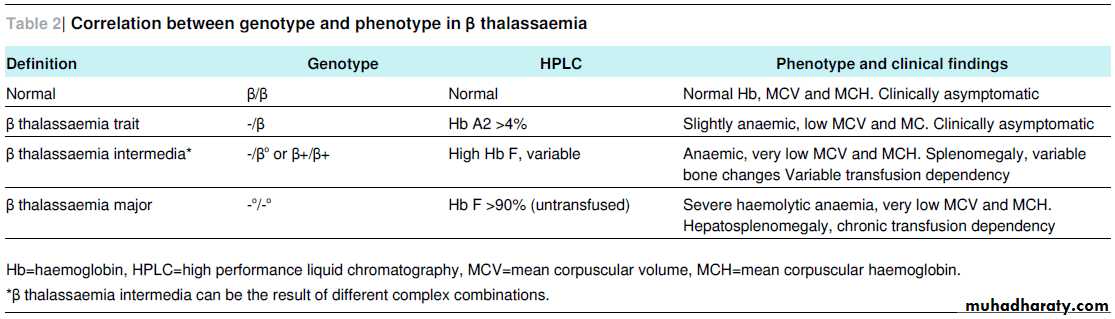
(phenotype) or genotype (type of mutation), which can be quite variable (tables 1⇓ and 2⇓).
Which populations are at risk?
β thalassaemia is prevalent in areas around the Mediterranean,in the Middle East, in Central, South, and South East Asia, and in Southern China; α thalassaemia is prevalent in South East. Asia, Africa, and India. Population cohort studies estimated that
worldwide 1.5% (80-90 million people) are carriers of β thalassaemia and 5% are carriers of αthalassaemia.
The high prevalence of α and β thalassaemia genotypes in communities with endemic Plasmodium falciparum malaria has led to a theory that the thalassaemia gene mutations represent a mechanism of evolutionary protection. Recent migrations of populations at
risk to non-endemic countries has resulted in increasing
prevalence of thalassaemia gene mutations in all parts of the world.
What are the different forms of α thalassaemia?
Normal individuals have two active α globin genes on both copies of chromosome 16. Table 1⇓ summarises the phenotypes seen in α thalassemia, showing how different clinical presentations are associated with the number of affected α globin genes.Homozygosity for the South East Asian α thalassaemia deletion,a condition in which the expression of both α globin genes on both alleles is completely absent (-,-/-,-), is one of the most common causes of hydrops fetalis, an accumulation of fluid in two or more fetal compartments that occurs secondary to
anaemia and fetal heart failure. Fetal haemoglobin (haemoglobin F) (α2/γ2) cannot be synthesised and haemoglobin Bart’s (γ4) becomes the major haemoglobin fraction in fetal life.
Since this homotetramer is not functional, the only oxygen transporting protein present is haemoglobin Portland, which is switched off early in embryonic development. Profound anaemia, organ damage, and in most cases perinatal death is the result. The
incidence of hydrops fetalis among births is highest in China (0.23%).
Haemoglobin H disease is caused by dysfunction of three of the four α globin genes (-,-/-,α). An excess of unpaired haemoglobin γ chains in utero results in high concentrations of haemoglobin Bart’s at birth and formation of the highly unstable andnon-functional haemoglobin H (β4) later on.
The newborn may present with moderate haemolytic anaemia and hepatosplenomegaly. A longitudinal study in 60 adult patients showed that haemoglobin H was not associated with an increased rate of severe anaemia and infections and was managed without blood transfusions.
Individuals with one or two non-functional α globin genes are usually clinically asymptomatic.
What are the different forms of β thalassaemia?
Normal individuals have one active β globin gene on both copies of chromosome 11. More than 200 mutations of these genes have been described, with different clinical consequences. The correlation between genotype and phenotype in β thalassaemiais summarised in table 2.⇓ Carriers of β thalassaemia (in whom the condition is called β thalassaemia trait or minor) are generally healthy individuals, while homozygosity or compound heterozygosity (a different
mutation in each allele) will result in a severe condition.
β thalassaemia major
Infants with β thalassaemia major become progressively pale and develop feeding problems and irritability from 3 months of age. Massive hepatosplenomegaly results from haemolysis and extramedullary haematopoiesis.If not treated, children will be affected by the complications of severe haemolytic anaemia and will develop growth retardation, poor muscular development, short deviated legs, a tendency to pathological fractures, and frontal bossing and maxillary prominence (facies thalassaemia)
secondary to bone marrow expansion—the body’s reaction to the anaemia is autogenous, although ineffective, erythropoiesis.
The occurrence of growth retardation and facies thalassaemia depends on the severity and duration of the anaemia. Increased gastrointestinal iron absorption may lead to iron overload in almost all organs, most importantly the liver and the heart.
Without proper treatment life expectancy is less than 5-10 years, with death usually due to cardiac failure.
About 50% of all cases of severe β thalassaemia phenotype in Southeast Asia are caused by coinheritance of β thalassemia and haemoglobin E (haemoglobin E β thalassaemia). This form
of the condition shows great variation in clinical severity,
ranging from an asymptomatic to a transfusion dependent phenotype.
β thalassaemia intermedia
β thalassaemia intermedia is characterised by later and milder onset of the clinical symptoms of β thalassaemia major. By definition, patients maintain a haemoglobin concentration above 70 g/L, often at the cost of intense bone marrow hyperplasiaand splenomegaly. Increase of gastrointestinal iron absorption may result in non-transfusional iron overload in the liver but, by contrast with β thalassaemia major, not in the heart.
How can thalassaemia be diagnosed?
Symptoms of haemolytic anaemia—paleness andhepatosplenomegaly—directly from birth in α thalassaemia, or from several months after birth in β thalassaemia, indicate asevere form of the disease, especially if microcytic anaemia is present, and second line testing has to be performed immediately.
The key sign of minor, intermediate, or major thalassaemia is microcytic, hypochromic red blood cells (mean corpuscular volume <70 fL), not responding to iron supplementation. It is therefore important to evaluate the effect of iron supplementation
by testing ferritin concentration and mean corpuscular volume in order to discriminate non-adherent patients from those in whom second line testing is indicated, independent of their apparent ethnic background.
Second line testing consists of separation and measurement of the haemoglobin fractions with high performance liquid chromatography or capillary electrophoresis. β thalassaemia major shows no or very low haemoglobin A and persistent high
haemoglobin F. In β thalassaemia trait, the haemoglobin A2 fraction is slightly raised (>4%).14
Haemoglobin H disease (-,-/-,α) presents with a variable degree of paleness at birth and laboratory testing shows microcytic anaemia.
Second line testing shows about 20% haemoglobin
Bart’s (γ4) in the neonatal period and variable amounts of haemoglobin H (β4) later in life.
In patients with mild microcytic anaemia, in the absence of iron deficiency and normal haemoglobin fractions in second line testing, the most probable diagnosis will be a mild form of αthalassaemia, to be confirmed by DNA analysis.
Third line testing at DNA level by standard Gap-PCR
technologies or direct DNA sequencing is only available in specialised laboratories. DNA analysis should be offered to identify and confirm couples at risk, in prenatal testing and in preimplantation genetic diagnosis.
Can thalassaemia be prevented?
Prevention of thalassemia is based on public awareness of the disease, detection of carriers, genetic counselling, and prenatal testing. In several countries, including the UK, antenatal and neonatal screening programmes for haemoglobinopathy enable early recognition of couples at risk and readily diagnose children with severe forms of α and β thalassaemia major (www.sct.screening.nhs.uk).Carriers of α thalassaemia with deletions on
the same chromosome (-,-/α,α) should be offered genetic counselling to estimate the risk of hydrops fetalis or a child with haemoglobin H disease.6 Testing and counselling should also be offered to all first degree relatives of a patient diagnosed with thalassaemia.How are patients with thalassaemia treated?
Asymptomatic carriersCarriers require no specific treatment. However, they should be protected from detrimental iron supplementation given merely on the basis of microcytosis. Iron supplementation should be
given only after confirmation of iron deficiency.
Thalassaemia intermedia
Patients with thalassaemia intermedia or haemoglobin H disease need to be closely monitored for progression of complications induced by chronic haemolytic anaemia. Symptoms usually develop when the haemoglobin level is sustained at below 70 g/L.Occasional blood transfusion may be required during
periods of rapid growth, infection-associated aplastic or hyperhaemolytic crises and pregnancy. Current indications for transfusion in thalassaemia intermedia are summarised in boxIndications for regular transfusion include growth
impairment and skeletal deformities. If hypersplenism develops, splenectomy may be considered, although this intervention carries severe risks of life threatening infections, pulmonary hypertension, and thrombosis.
Thalassaemia major
In 2008 the Thalassaemia International Federation published
guidelines for the clinical management of thalassaemia major.
The cornerstones of treatment are:
Regular hypertransfusion to maintain a haemoglobin level higher than 95 g/L
• Iron chelation to prevent overload syndrome
• Care by a multidisciplinary team (haematologist, paediatrichaematologist, specialised nurse, social worker,
psychologist, genetic counseller, cardiologist, and
hepatologist).
Survival data derived from large cohort studies have shown that patients undergoing regular transfusion but not chelation can be expected to reach 10-25 years of age, dying of cardiac iron
overload.
Patients receiving optimal transfusion and chelation from an early age, however, have a substantially increased survival of more than 40 years. These data are derived from The UK Thalassaemia Register, which records births, deaths, and selected clinical data of patients with thalassaemia who are resident in the UK.
Considerations regarding hypertransfusion
treatmentHypertransfusion decreases autogenous, ineffective
erythropoiesis and haemolytic anaemia. However, an adverse effect of this treatment is iron overload, because each unit of red blood cells adds 200 mg of iron, which can only be slowly removed from the body.
The complications of iron overload are
cardiomyopathy and liver cirrhosis. Furthermore, transfusional iron overload affects endocrine organs (hypothalamus, pituitary, gonads, pancreas, thyroid and parathyroid glands), which in
combination with the impaired general condition of the patient results in growth impairment, delayed or absent puberty, infertility, and diabetes mellitus.
Cardiac failure is the most common fatal complication in patients with thalassaemia major
treated with chronic transfusion. The risk of transmission of viral infections (HIV, hepatitis B, and hepatitis C) remains aserious problem in many countries. To reduce the risk of allo-immunisation, perform extended red blood cell antigenphenotyping before starting transfusions. Leucocyte depleted transfused red blood cells (<1x106 leucocytes per unit) reduce adverse reactions attributed to contaminating white cells.
How should iron overload be monitored?
It is important to evaluate iron concentration and function in the heart and the liver. Disparity between liver and cardiac iron loading may exist because of differences in the mechanisms and kinetics of iron uptake and clearance between the organs.Although several observational studies have shown an between high concentrations of ferritin in serum and early death, the prediction of iron loading (especially cardiac iron) from serum ferritin can be unreliable. In an intervention study, patients with ferritin levels below 2500 μg/L over a period
of more than a decade had significantly reduced risks of cardiac disease and death, compared with those who had ferritin levels of more than 2500 μg/L.21
Iron concentration in the liver can be determined by biopsy. Adiagnostic study found that liver biopsy accurately predicts total body iron stores and is considered to be the gold standard test for hepatic iron overload. Two non-invasive methods based
on magnetic resonance imaging are available to quantify hepatic iron concentration: the T2 and T2 star (T2*) techniques. Adiagnostic study showed, for a 1 or 1.5-T MRI, a sensitivity of 92% and a specificity of 57% for the absence of overload, out or diagnose high iron overload in only 75% of patients.
Compromised left ventricular ejection fraction (LVEF) is a late sign of cardiomyopathy and is therefore not of use in the prevention and early detection of cardiac iron overload.
However, estimation of myocardial iron with T2* MRI can be used to select patients with an increased chance of cardiac failure before a fall in LVEF occurs.
Cardiac iron levels are inversely related to myocardial T2* values. Values less than 20 ms indicate detectable cardiac iron and are associated with an increased chance of compromised left ventricular function. The relative risk of heart failure with cardiac T2* values <10 ms is 160 (95% confidence interval 39 to 653) compared with those with a value of >10 ms, according to a prospective follow-up study in 652 patients with thalassaemia major (cardiac T2* was <10 ms in 98% of patients who developed heart failure).
How should iron overload be prevented
and treated?
Experts recommend that iron overload be treated when serum ferritin levels exceed 1000 μg/L, which will occur after 10 to 20 red cell transfusions.
In the early 1960s the use of deferoxamine was introduced. This hexadentate requires subcutaneous infusion over 8-12 hours on five to seven nights per week, and leads to decreased serum ferritin concentrations as a result of urinary and faecal iron excretion.
In the late 1980s the oral chelator deferiprone became available, a bidentate leading to urinary iron excretion that can be administered three times daily. In 2006 the first reports were
published on oral deferasirox, a tridentate giving rise to faecal iron excretion and administered once daily.
A recent Cochrane Review found little consistency between the outcomes of all studies that have examined the efficacy and safety of deferiprone in comparison to deferoxamine, and no
recommendations could be made.25 The findings of some studies suggest that deferiprone is more effective than deferoxamine in removing cardiac iron.
Galanello recommends treatment with deferiprone in combination with deferoxamine in patients with
severe cardiac iron overload.26 Deferasirox is a very effective chelator, but it is not superior to the other chelating drugs. In a recently published study, long term follow-up of both adults and children showed good safety and efficacy of deferasirox in removing liver iron and reversed or stabilised liver fibrosis in
83% of the patients.
Furthermore, a longitudinal study in 39 patients showed that continuous treatment with deferasirox for two years, with atarget dose of 40 mg/kg per day, continued to remove iron from
the heart in patients with β thalassemia major and mild,moderate, or severe cardiac siderosis.
Adherence to chelation therapy is cumbersome but essential; survival is poor in non-adherent patients. Practical and psychological support is needed, therefore, to ensure optimal long term adherence. If subcutaneous treatment is too burdensome for a patient, oral therapy is usually first choice.
When serious adverse effects, progressive iron overload, or non-adherence occur, deferoxamine can be restarted. Experts agree that the choice of iron chelator should be tailored to the
individual patient.
Alternative therapeutic options
Splenectomy can be considered if hypersplenism causes amarked increase in transfusion requirements. In general, it should be delayed for as long as possible, in order to prevent life threatening infections, pulmonary hypertension and thrombo-embolic complications.
Hydroxyurea, a cytotoxic drug, may increase the expression of γ chains (haemoglobin F) and enables the neutralisation of excess α chains, which could potentially correct ineffective erythropoiesis.
Is there a potential cure?
The only potential cure for β thalassaemia is haematopoietic stem cell transplantation. In this procedure, autologous hematopoiesis is eradicated through chemotherapy. Additionaleradication of residual immune cells, before the infusion of new hematopoietic stem cells, will render the patient completely immunocompromised.
Infections will be a major hazard in addition to the side effects of chemotherapy.
The success of stem cell transplantation depends on the amount of erythrocyte transfusions received and the severity of iron overload. The recent developments of new chemotherapeutic conditioning regimens and improved supportive care have decreased the rate of fatal complications considerably.The potential for success is furthermore determined by HLA matching between the donor stem cells and patient HLA. The preferred source of donor stem cells is an HLA identical family member. Stem cells derived from an unrelated matched donor
have been shown to be a good alternative for stem cell transplantation in β thalassaemia.
Differences in HLA between patient and donor will determine the risk of either non-engraftment (donor cells will be rejectedby patient) or graft versus host disease (donor cells will attack the patient cells). A new strategy is the use of HAPLO identical stem cells derived from one of the patient’s parents. Donation
of highly purified stem cells will lower the risk of graft versus host disease. However, the risk of rejection is raised, immune reconstitution delayed, and therefore risk of infection increased.
Bacterial, fungal, or viral infections are all potentially lethal in the highly immunocompromised patient. The use of virus specific T cells to treat patients with viral reactivations makes HAPLO identical stem cell transplantation a more attractive alternative.
Despite the risks, hematopoietic stem cell transplantation remains a curative option for most thalassaemia patients who depend on transfusion, with overall survival of 90% and disease
free survival of 86% for a mean follow-up period of 15 years.
The younger the patient is at time of stem cell transplantation, the better the outcome. Neonatal screening programmes can identify patients with β thalassaemia major at a young age, creating the chance to perform a transplant before the
complications of the condition appear.
Summary points
The changing demographic features of thalassaemia, with its widely variable phenotypes, have implications for diagnosis, counselling, and management Carriers of thalassaemia require no specific treatment but should be protected from iron supplementation, which may be detrimental .Antenatal and neonatal screening for thalassaemia may reduce the number of severely affected children .
Monitoring iron overload and chelation therapy in transfusion dependent thalassaemia patients is essential to prevent fatal cardiomyopathy
and liver cirrhosis .The only potential cure for β thalassaemia is hematopoietic stem cell transplantation.
Box 1 Indications for transfusion in β thalassaemia intermedia
Haemoglobin concentration <50 g/LFalling haemoglobin level with profound enlargement of the spleen .Growth failure or poor performance at school .Diminished exercise tolerance
Failure of secondary development in parallel with bone age
Severe bone changes
Pregnancy
Infection
Other specific complications: heart failure, pulmonary complications, hypertension, thromboembolic disease, leg ulcers, priapism.
Box 2 Components of regular treatment for β thalassaemia major
Red blood cell transfusions to maintain haemoglobin level higher than 95 g/LPrevention of complications of a chronic transfusion programme: allo-immunisation, iron overload, viral infections (hepatitis C and B, HIV, parvovirus B19)
Prompt treatment of all infections
Splenectomy (hypersplenism)
Hydroxyurea
Active donor search for haematopoietic stem cell transplantation
Endocrinological, skeletal, hepatic, and cardiac monitoring
Chelation for patients with iron overload.
Questions for future research
• Can we identify, at an early stage, patients with thalassaemia intermedia at risk of late onset complications?• Will newly developed iron chelation supported by hepcidin agonists be effective?
• Will better understanding of the kinetics of iron metabolism, iron overload, and chelation improve therapeutic strategies?
• Will pharmacological approaches to increase the expression of fetal haemoglobin or correct the mutated β globin mRNA prevent or
limit the need for blood transfusion?
• Is gene therapy the ultimate answer in thalassaemia?



