
Dyslipidemia
DISORDERS OF LIPIDS AND LIPOPROTEINSد. حسين محمد جمعه
اختصاصي الامراض الباطنة
البورد العربي
كلية طب الموصل
2011
Plasma lipoproteins are composed of a hydrophobic lipids core of TG and cholesterol ester, enveloped by a surface coat of hydrophilic lipids ,phospholipid, unesterified ("free") cholesterol, and special proteins called apolipoproteins (or apoproteins).
Lipoproteins are large macromolecular complexes that transport hydrophobic lipids (primarily triglycerides, cholesterol, and fat-soluble vitamins) through body fluids (plasma, interstitial fluid, and lymph) to and from tissues.
Understanding the Essentials of Blood Lipid Metabolism: Lipid Terminology
Figure 16.13 Structure of lipoproteins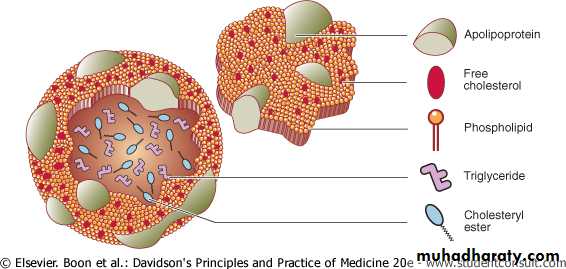
Structure of lipoproteins.
The term "lipoprotein" refers to this unique combination of "lipid and protein."
Lipoproteins play an essential role in the absorption of dietary cholesterol, long-chain fatty acids, and fat-soluble vitamins; the transport of triglycerides, cholesterol, and fat-soluble vitamins from the liver to peripheral tissues; and the transport of cholesterol from peripheral tissues to the liver.
The five main classifications of lipoproteins according to its density, lipid composition, and the apolipoproteins on the surface of the particle:are
chylomicrons,
very low-density lipoprotein cholesterol (VLDL), intermediate-density lipoprotein cholesterol (IDL), low-density lipoprotein cholesterol (LDL), and high-density lipoprotein cholesterol (HDL).
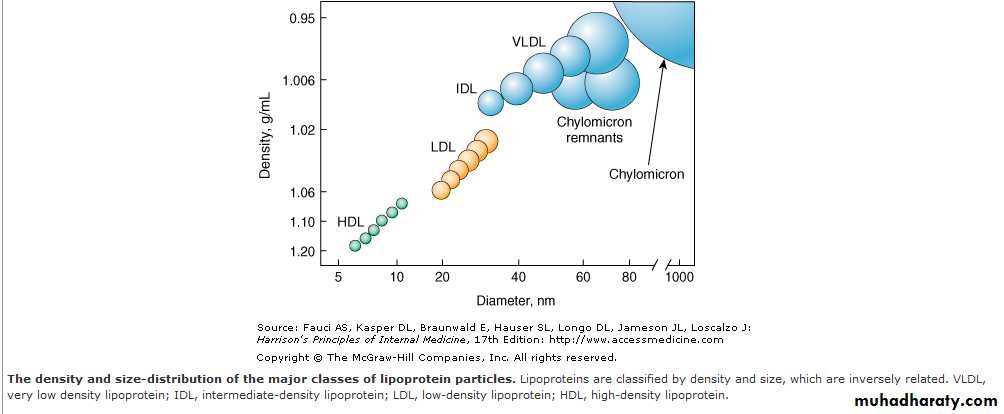
The density of a lipoprotein is determined by the amount of lipid per particle.
HDL is the smallest and most dense lipoprotein, whereas chylomicrons and VLDLs are the largest and least dense lipoprotein particles.Most plasma triglyceride is transported in chylomicrons or VLDLs, and
most plasma cholesterol is carried as cholesteryl esters in LDLs and HDLs.
Endogenous lipid
In the fasting state, the liver is the major source of plasma lipids .The liver may acquire lipids by uptake, synthesis or conversion from other macronutrients. These lipids are transported to other tissues by secretion of TG-rich very low-density lipoproteins (VLDL), which differ from chylomicrons in that they contain full-length Apo B100. Hydrolysis of VLDL TG releases fatty acids to tissues and converts VLDL into 'remnant' particles, (IDL). Most IDL are rapidly cleared by LDL receptors in the liver, but some are processed by hepatic lipase, which converts the particle to an LDL.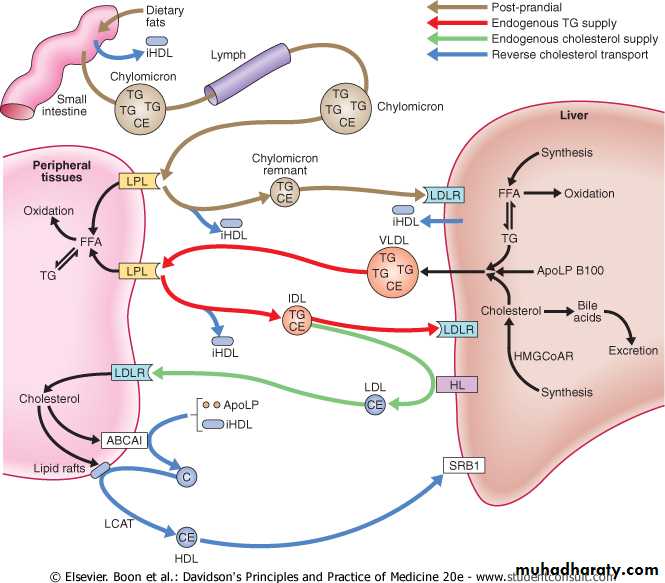
Pathways of lipid transport are shown; in addition cholesterol ester transfer protein exchanges triglyceride and cholesterol ester between VLDL/chylomicrons and HDL/LDL, and free fatty acids released from peripheral lipolysis can be taken up in the liver.
(ABCA1, ABCG1 = ATP-binding cassette A1, G1; Apo = apolipoprotein; BA = bile acids; C = cholesterol; CE = cholesterol ester; FFA = free fatty acids; HDL = mature high-density lipoprotein; iHDL = immature high-density lipoprotein; HL = hepatic lipase; HMGCoAR = hydroxy-methyl-glutaryl-coenzyme A reductase; IDL = intermediate-density lipoprotein; LCAT = lecithin cholesterol acyl transferase; LDL = low-density lipoprotein; LDLR = low-density lipoprotein receptor (Apo B100 receptor); LPL = lipoprotein lipase; SRB1 = scavenger receptor B1; TG = triglyceride; VLDL = very low-density lipoprotein)
Absorption, transport and storage of lipids.
Peripheral tissues are further guarded against excessive cholesterol accumulation by high-density lipoproteins . Lipid-poor Apo A1 (derived from the liver, intestine and the outer layer of chylomicrons and VLDL) accepts cellular cholesterol and phospholipid from a specific membrane transporter known as the ATP-binding cassette A1 transporter (ABC A1). This produces small HDLs that are able to accept more free cholesterol from cholesterol-rich regions of the cell membrane via another membrane transporter (ABC G1).
Reverse cholesterol transport
The cholesterol that has been accepted by these small HDLs is esterified by lecithin cholesterol acyl transferase (LCAT), thus maintaining an uptake gradient and remodelling the particle into a mature spherical HDL.
These HDL release their cholesterol to the liver and other cholesterol-requiring tissues via the scavenger receptor B1 (SRB1). The cholesterol ester transfer protein (CETP) in plasma allows transfer of cholesterol from HDL or LDL to VLDL or chylomicrons in exchange for TG.
When TG is elevated, the action of CETP may reduce HDL cholesterol and remodel LDL into 'small, dense' LDL particles that appear to be more atherogenic in the blood vessel wall. Animal species that lack CETP are resistant to atherosclerosis, suggesting that this process may have a detrimental effect on progression of cardiovascular disease.
Plasma lipoprotein levels are major modifiable risk factors for cardiovascular disease. Increased levels of atherogenic lipoproteins (especially LDL, but also IDL, lipoprotein (a) and possibly chylomicron remnants) contribute to the development of atherosclerosis . Plasma cholesterol and TG are clinically important mainly because they are major treatable risk factors for cardiovascular disease, whilst severe hypertriglyceridaemia also predisposes to acute pancreatitis.
Lipids and cardiovascular disease
Increased plasma concentration and reduced diameter favour subendothelial accumulation of these lipoproteins.Following oxidation, these Apo B-containing lipoproteins are no longer cleared by normal mechanisms.
They trigger a self-perpetuating inflammatory response during which they are taken up by macrophages to form foam cells, a hallmark of atherosclerotic lesions.
Conversely, HDL removes cholesterol from the tissues to the liver, where it is metabolised and excreted in bile.
HDL may also counteract some components of the inflammatory response, such as the expression of vascular adhesion molecules by the endothelium.
Consequently, low HDL cholesterol levels, which are often associated with triglyceride elevation, also predispose to atherosclerosis.
Lipid measurement
Abnormalities of lipid metabolism most commonly come to light following routine blood testing. Measurement of plasma cholesterol alone is not sufficient for comprehensive assessment.
Levels of (TC), (TG) and (HDL-C) need to be obtained after a 12-hour fast to permit accurate calculation of LDL cholesterol (LDL-C) according to the Friedewald formula (LDL-C = TC - HDL-C - (TG/2.2) mmol/L).
(Before the formula is applied, lipid levels in mg/dL can be converted to mmol/L by dividing by 38 for cholesterol and 88 for triglycerides.) The formula becomes unreliable when TG levels exceed 4 mmol/L (350 mg/dL).
However, non-fasting samples are often used to guide therapeutic decisions since they are unaffected in terms of TC and measured LDL-C, albeit that they differ from fasting samples in terms of TG, HDL-C and, to some extent, calculated LDL-C.
In the laboratory, total cholesterol, TG, and HDL are measured directly, from which a calculated estimation of VLDL and LDL cholesterol are derived. These measurements of VLDL and LDL are based on a fasting TG level and are valid only when the TG level is less than 400 mg/dL (5 mmol/L). This explains the requirement for a fasting blood sample (overnight fast of 12 hours) to determine blood lipid levels.
Direct measurement of VLDL and LDL is also possible; however, due to their high cost and technical complexity, these are performed primarily in reference laboratories.
Hypertriglyceridaemia interferes with the serum amylase assay and can produce a falsely low result—serum amylase can seem to be within normal limits despite clinical and radiological evidence of acute pancreatitis. Urine amylase to creatinine ratio can be measured to diagnose acute pancreatitis, and the result of this test is less likely to be affected by hypertriglyceridaemia.
Alternatives are the removal of lipids before serum amylase measurement by using ultracentrifugation..
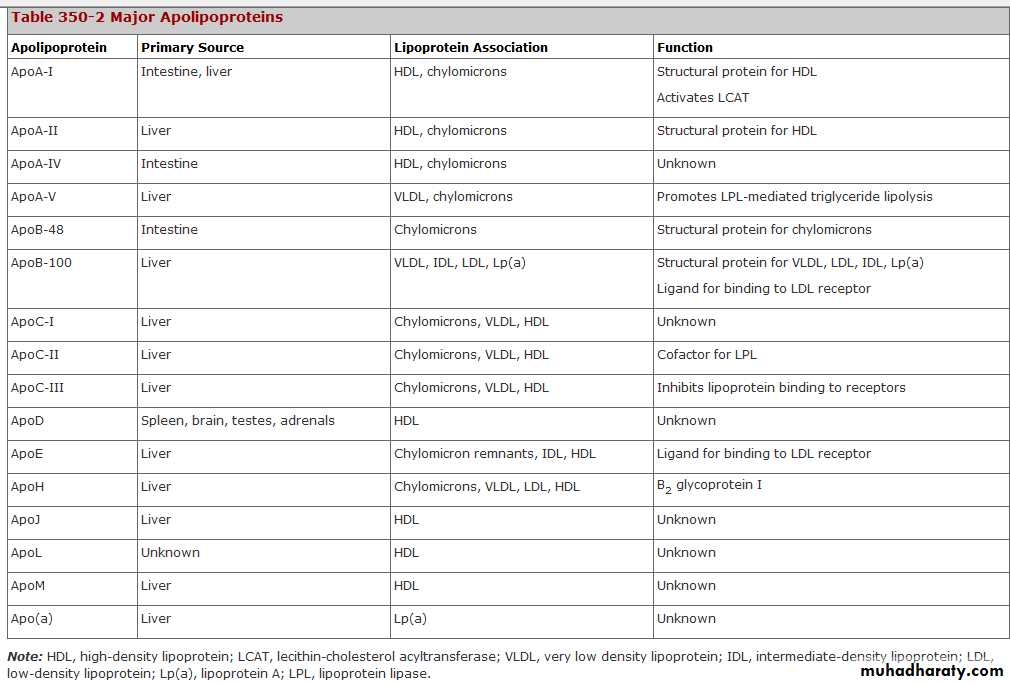
The proteins associated with lipoproteins, called apolipoproteins, are required for the assembly, structure, and function of lipoproteins. Apolipoproteins activate enzymes important in lipoprotein metabolism and act as ligands for cell surface receptors.
ApoA-I, which is synthesized in the liver and intestine, is found on virtually all HDL particles. ApoA-II is the second most abundant HDL apolipoprotein and is on approximately two-thirds of all HDL particles.
ApoB is the major structural protein of chylomicrons, VLDLs, IDLs, and LDLs; one molecule of apoB, either apoB-48 (chylomicron) or apoB-100 (VLDL, IDL or LDL), is present on each lipoprotein particle.
Consideration must be given to confounding factors, such as recent illness, after which cholesterol levels temporarily decrease in proportion to severity.
Results that will affect major decisions, such as initiation of drug therapy, should be confirmed with a repeat measurement.
Cholesterol and retinol are esterified (by the addition of a fatty acid) in the enterocyte to form cholesteryl esters and retinyl esters, respectively. Longer-chain fatty acids (>12 carbons) are incorporated into triglycerides and packaged with apoB-48, cholesteryl esters, retinyl esters, phospholipids and cholesterol to form chylomicrons.
Nascent chylomicrons are secreted into the intestinal lymph and delivered via the thoracic duct directly to the systemic circulation, where they are extensively processed by peripheral tissues before reaching the liver. The particles encounter lipoprotein lipase (LPL), which is anchored to proteoglycans that decorate the capillary endothelial surfaces of adipose tissue, heart and skeletal muscle .
The triglycerides of chylomicrons are hydrolyzed by LPL, and free fatty acids are released. ApoC-II, which is transferred to circulating chylomicrons from HDL, acts as a cofactor for LPL in this reaction. The released free fatty acids are taken up by adjacent myocytes or adipocytes and either oxidized to generate energy or reesterified and stored as triglyceride.
Some of the released free fatty acids bind albumin before entering cells and are transported to other tissues, especially the liver. The chylomicron particle progressively shrinks in size as the hydrophobic core is hydrolyzed and the hydrophilic lipids (cholesterol and phospholipids) and apolipoproteins on the particle surface are transferred to HDL, creating chylomicron remnants. Chylomicron remnants are rapidly removed from the circulation by the liver through a process that requires apoE as a ligand for receptors in the liver.
Consequently, few, if any, chylomicrons or chylomicron remnants are present in the blood after a 12-h fast, except in patients with disorders of chylomicron metabolism.
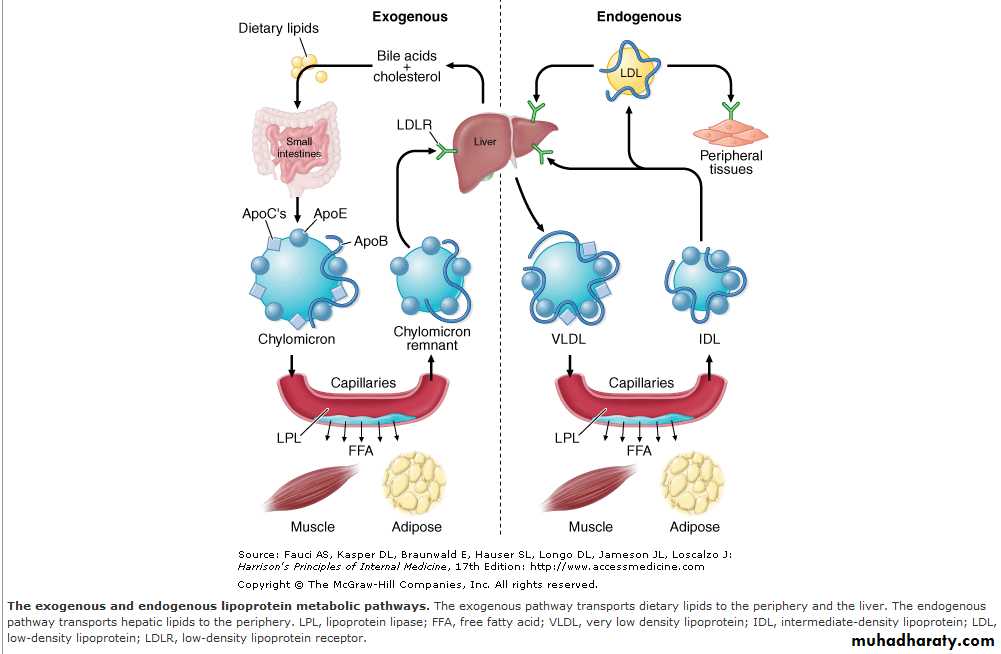
Transport of Hepatic Lipids (Endogenous Pathway)
The endogenous pathway of lipoprotein metabolism refers to the hepatic secretion of apoB-containing lipoproteins and their metabolism .VLDL particles resemble chylomicrons in protein composition but contain apoB-100 rather than apoB-48 and have a higher ratio of cholesterol to triglyceride (~1 mg of cholesterol for every 5 mg of triglyceride).
The triglycerides of VLDL are derived predominantly from the esterification of long-chain fatty acids in the liver.
The packaging of hepatic triglycerides with the other major components of the nascent VLDL particle (apoB-100, cholesteryl esters, phospholipids, and vitamin E) requires the action of the enzyme microsomal triglyceride transfer protein (MTP).
After secretion into the plasma, VLDL acquires multiple copies of apoE and apolipoproteins of the C series by transfer from HDL. As with chylomicrons, the triglycerides of VLDL are hydrolyzed by LPL, especially in muscle and adipose tissue.
After the VLDL remnants dissociate from LPL, they are referred to as IDLs, which contain roughly similar amounts of cholesterol and triglyceride. The liver removes approximately 40–60% of IDL by LDL receptor–mediated endocytosis via binding to apoE. The remainder of IDL is remodeled by hepatic lipase (HL) to form LDL.
During this process, most of the triglyceride in the particle is hydrolyzed, and all apolipoproteins except apoB-100 are transferred to other lipoproteins. The cholesterol in LDL accounts for over half of the plasma cholesterol in most individuals. Approximately 70% of circulating LDL is cleared by LDL receptor–mediated endocytosis in the liver.
Lipoprotein(a) [Lp(a)] is a lipoprotein similar to LDL in lipid and protein composition, but it contains an additional protein called apolipoprotein(a) [apo(a)]. Apo(a) is synthesized in the liver and attached to apoB-100 by a disulfide linkage. The major site of clearance of Lp(a) is the liver, but the uptake pathway is not known.
HDL Metabolism and Reverse Cholesterol Transport
All nucleated cells synthesize cholesterol, but only hepatocytes and enterocytes can effectively excrete cholesterol from the body, into either the bile or the gut lumen.In the liver, cholesterol is excreted into the bile, either directly or after conversion to bile acids.
Cholesterol in peripheral cells is transported from the plasma membranes of peripheral cells to the liver and intestine by a process termed "reverse cholesterol transport" that is facilitated by HDL .
Cholesterol in peripheral cells is transported from the plasma membranes of peripheral cells to the liver and intestine
by a process termed "reverse cholesterol transport" that is facilitated by HDL .
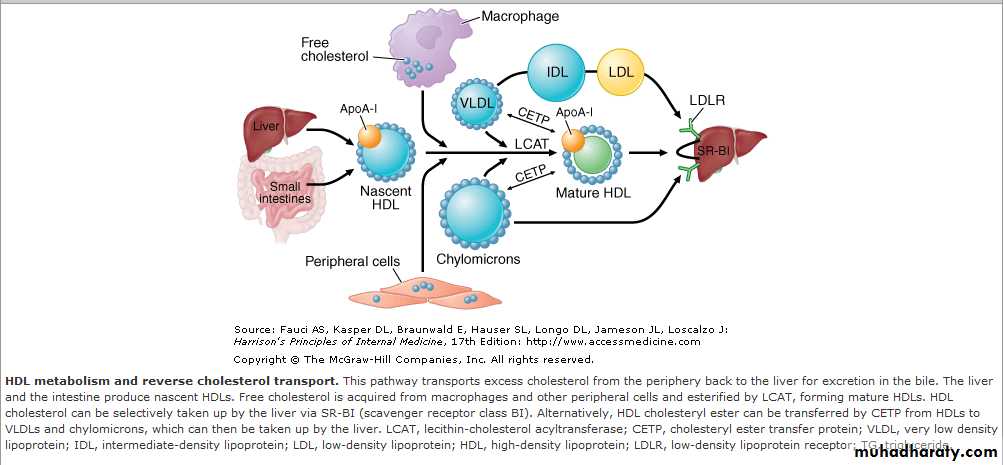
Nascent HDL particles are synthesized by the intestine and the liver.
Newly secreted apoA-I rapidly acquires phospholipids and unesterified cholesterol from its site of synthesis (intestine or liver) via efflux promoted by the membrane protein ATP-binding cassette protein A1 (ABCA1). This process results in the formation of discoidal HDL particles, which then recruit additional unesterified cholesterol from the periphery.Within the HDL particle, the cholesterol is esterified by lecithin-cholesterol acyltransferase (LCAT), a plasma enzyme associated with HDL, and the more hydrophobic cholesteryl ester moves to the core of the HDL particle. As HDL acquires more cholesteryl ester it becomes spherical, and additional apolipoproteins and lipids are transferred to the particles from the surfaces of chylomicrons and VLDLs during lipolysis.
Transport of Dietary Lipids (Exogenous Pathway)
The exogenous pathway of lipoprotein metabolism permits efficient transport of dietary lipids .Dietary triglycerides are hydrolyzed by lipases within the intestinal lumen and emulsified with bile acids to form micelles.Dietary cholesterol, fatty acids, and fat-soluble vitamins are absorbed in the proximal small intestine.
Lipid measurements are usually performed for the following reasons:
screening for primary or secondary prevention of cardiovascular diseaseinvestigation of patients with clinical features of lipid disorders .
testing relatives of patients with one of the single gene defects causing dyslipidaemia.
Lipid measurements
Causes of secondary hyperlipidaemia
Secondary hypercholesterolaemiaModerately common
• Hypothyroidism• Pregnancy
• Cholestatic liver disease
• Drugs (diuretics, ciclosporin, corticosteroids, androgens)
Diabetes mellitus (type 2)
Chronic renal disease
Abdominal obesity
Excess alcohol
Hepatocellular disease
Drugs (β-blockers, retinoids, corticosteroids)
• Secondary hypertriglyceridaemia
Common• Less common
Nephrotic syndrome
Anorexia nervosa
Porphyria
Hyperparathyroidism
Once secondary causes are excluded, primary lipid abnormalities may be diagnosed.
The Fredrickson classification (types I-V) adds little to clinical decision-making.Alternatively, primary lipid abnormalities can be classified according to the predominant lipid problem:
• hypercholesterolaemia,
• hypertriglyceridaemia or
• mixed hyperlipidaemia
primary lipid abnormalities
Although single gene disorders are encountered in all three categories, the most common cause is an interaction between numerous genetic and environmental factors (i.e. 'polygenic'). Clinical consequences of dyslipidaemia vary somewhat between these categories .
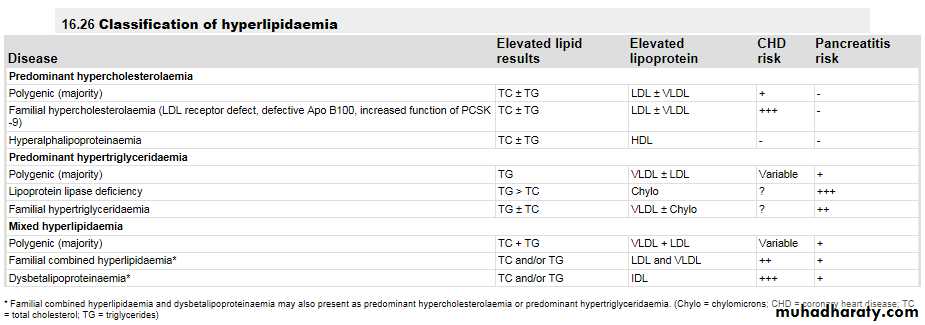
Predominant hypercholesterolaemia
Polygenic hypercholesterolaemia is the most common cause of a mild to moderate increase in LDL-C .Physical signs such as corneal arcus and xanthelasma may be found in this as well as other forms of lipid disturbance .The risk of cardiovascular disease is proportional to the degree of LDL-C elevation, but is modified by other major risk factors, particularly low HDL-C.Predominant hypercholesterolaemia
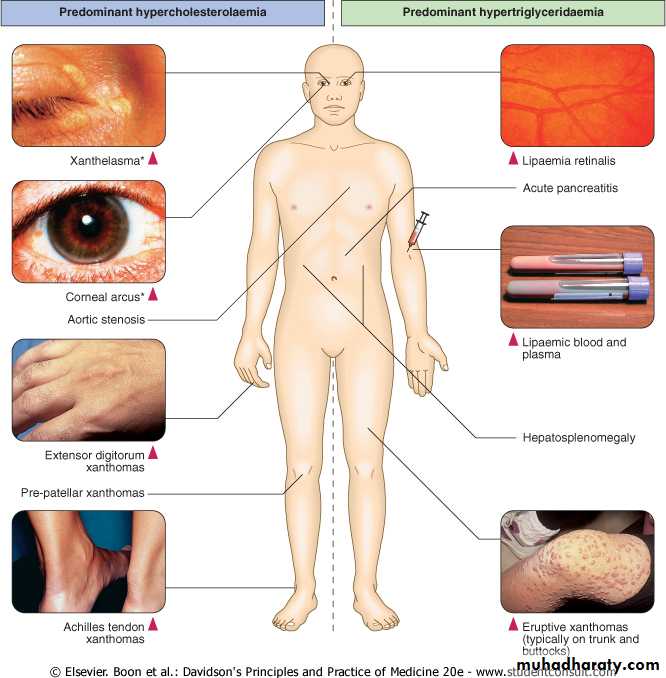
Clinical manifestations of hyperlipidaemia. *Note that xanthelasma and corneal arcus may be non-specific, especially in later life.
Hyperalphalipoproteinaemia refers to increased levels of HDL-C. In the absence of an increase in LDL-C, this condition does not cause cardiovascular disease, so it should not be regarded as pathological
Predominant hypertriglyceridaemia
Polygenic hypertriglyceridaemia is the most common primary cause of TG elevation. It also commonly occurs secondary to excess alcohol, medications, type 2 diabetes, impaired glucose tolerance, central obesity or other manifestations of the insulin resistance syndrome.It is difficult to define quantitatively the distinction between predominant hyperlipidaemias and mixed hyperlipidaemia. The term 'mixed' usually implies the presence of hypertriglyceridaemia as well as an increase in LDL or IDL.
Mixed hyperlipidaemia
Primary mixed hyperlipidaemia is usually polygenic and, like predominant hypertriglyceridaemia, often occurs in association with type 2 diabetes, impaired glucose tolerance, central obesity or other manifestations of the insulin resistance syndrome .Both components of mixed hyperlipidaemia may contribute to the risk of cardiovascular disease.
Dysbetalipoproteinaemia (also referred to as type 3 hyperlipidaemia, broad-beta dyslipoproteinaemia or remnant hyperlipidaemia) involves accumulation of roughly equimolar levels of cholesterol and TG. It is caused by homozygous inheritance of the Apo E2 allele, which is the isoform least avidly recognised by the LDL receptor.
Management of dyslipidaemia
Lipid-lowering therapies have a key role in the secondary and primary prevention of cardiovascular diseases .Assessment of absolute risk,
treatment of all modifiable risk factors and optimisation of lifestyle, especially diet and exercise, are central to management in all cases.
Patients with the greatest absolute risk of cardiovascular disease will derive the greatest benefit from treatment.
In general, patients who already have cardiovascular disease, diabetes mellitus, chronic renal impairment or an absolute risk of cardiovascular disease of greater than 20% in the ensuing 10 years are arbitrarily regarded as having sufficient risk to justify drug treatment.
Public health organisations also recommend target levels for patients receiving drug treatment. High-risk patients should aim for HDL-C > 1 mmol/L (38 mg/dL) and fasting TG < 2 mmol/L (approximately 180 mg/dL), whilst target levels for LDL-C have been reduced from 2.5 to 2.0 mmol/L (76 mg/dL) or less. In general, total cholesterol should be < 5 mmol/L (190 mg/dL) during treatment, and < 4 mmol/L (approximately 150 mg/dL) in high-risk patients and in secondary prevention of cardiovascular disease.
Non-pharmacological management
Patients with lipid abnormalities should receive medical advice and, if necessary, dietary counselling to:’reduce intake of saturated and trans-unsaturated fat to less than 7-10% of total energy reduce intake of cholesterol to < 250 mg/day replace sources of saturated fat and cholesterol with alternative foods such as lean meat, low-fat dairy products, polyunsaturated spreads and low glycaemic index carbohydrates
reduce energy-dense foods such as fats and soft drinks, whilst increasing activity and exercise to maintain or lose weight.
increase consumption of cardioprotective and nutrient-dense foods such as vegetables, unrefined carbohydrates, fish, pulses, nuts, legumes, fruit etc.
adjust alcohol consumption, reducing intake if excessive or if associated with hypertension, hypertriglyceridaemia or central obesity
achieve additional benefits with supplementary intake of foods containing lipid-lowering nutrients such as n-3 fatty acids, dietary fibre and plant sterols. Even minor weight loss can substantially reduce cardiovascular risk, especially in centrally obese patients .
Very low-fat diets
Although very low-fat diets may indeed lower cholesterol levels, they are not recommended. Very low-fat diets usually allow less than 15% of total calories from fat.
In comparison, a cholesterol-reducing diet allows 25% to 35% of calories to come from total fat, with 7% from saturated fat.
A diet with less than 25% of its calories from fat can increase triglycerides and decrease HDL (good) cholesterol. Such a diet may deplete your body of other important nutrients and vitamins.
Pharmacological management
HMGCoA reductase inhibitors (statins)Statins inhibit cholesterol synthesis, thereby up-regulating activity of the LDL receptor. This increases clearance of LDL and its precursor, IDL, resulting in a secondary reduction in LDL synthesis.
Statins reduce LDL-C by up to 60%, reduce TG by up to 40% and increase HDL-C by up to 10%. There is clear evidence of protection against total and coronary mortality, stroke and cardiovascular events in high-risk patients .
Statins are generally well tolerated and serious side-effects are rare (well below 2%). Liver function test abnormalities and muscle problems such as myalgia, asymptomatic increase in creatine kinase (CK), myositis and, infrequently, rhabdomyolysis are the most common. Side-effects are more likely in patients who are elderly, debilitated or receiving other drugs that interfere with statin degradation, which usually involves cytochrome P450 3A4 or glucuronidation.
Benefits of treating patients with hypercholesterolaemia with statins
'Meta-analysis of major RCTs involving over 90 000 subjects receiving statins for an average of 5 years showedreduced mortality from
coronary artery disease, 19%
stroke, 17% and all causes, 12% (9-16%) per 1 mmol/L reduction in LDL-C.'
These inhibit the intestinal mucosal transporter NPC1L1 that absorbs dietary and biliary cholesterol. Depletion of hepatic cholesterol up-regulates hepatic LDL receptor activity. This mechanism of action is synergistic with the effect of statins. Monotherapy with the standard 10 mg/day dose reduces LDL-C by 15-20%.
Cholesterol absorption inhibitors, such as ezetimibe
Slightly greater (17-25%) incremental LDL-C reduction occurs when ezetimibe is added to statins. Ezetimibe is well tolerated, but its effect on cardiovascular disease endpoints is yet to be determined. Plant sterol-supplemented foods, which also reduce cholesterol absorption, lower LDL-C by 7-15%.
Bile acid sequestering resins, such as colestyramine, colestipol and colesevalam
These prevent the reabsorption of bile acids, thereby increasing de novo bile acid synthesis from hepatic cholesterol. As with ezetimibe, the resultant depletion of hepatic cholesterol up-regulates LDL receptor activity and reduces LDL-C in a manner that is synergistic with the action of statins. High doses (24 g/day colestyramine) can achieve substantial reductions in LDL-C and modest increases in HDL-C, but TG may rise.Resins are safe, but they may interfere with bioavailability of other drugs. Colesevalam may cause fewer gastrointestinal effects than older preparations. Development of specific inhibitors of the intestinal bile acid transporter may further improve tolerability of this class of agent.
Nicotinic acid (vitamin B3)
• In pharmacological doses, this reduces peripheral fatty acid release with the result that cholesterol and TG decline whilst HDL-C increases. Randomised clinical trials suggest a beneficial effect on atherosclerosis and cardiovascular events. Side-effects include flushing, gastric irritation, liver function disturbances, and exacerbation of gout and hyperglycaemia. Slow-release formulations and low-dose aspirin may reduce flushing. Combination therapy with the prostaglandin D2 receptor inhibitor laropiprant to further reduce flushing is being evaluated.Routine treatment of predominant hypercholesterolaemia generally requires continuation of diet plus the use of a statin in sufficient doses to achieve target LDL-C levels. Patients who do not reach LDL targets on the highest tolerated statin dose, or who are intolerant of statins, may receive ezetimibe, plant sterols, nicotinic acid or resins. Nicotinic acid is very effective in combination with a statin, but caution is required because the risk of side-effects is increased.
Fibrates
These stimulate peroxisome proliferator activated receptor (PPAR) alpha, which controls the expression of gene products that mediate the metabolism of triglyceride and HDL. As a result, synthesis of fatty acids, triglyceride and VLDL is reduced, whilst that of lipoprotein lipase, which catabolises TG, is enhanced. fibrates reduce TG by up to 50% and increase HDL-C by up to 20%, but LDL-C changes are variable.Predominant hypertriglyceridaemia
Fibrates are usually well tolerated, but share a similar side-effect profile to statins, including myalgia, myopathy and abnormal liver function tests. In addition, they may increase the risk of cholelithiasis and prolong the action of anticoagulants.Highly polyunsaturated long-chain n-3 fatty acids
Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) comprise approximately 30% of the fatty acids in fish oil. EPA and DHA are potent inhibitors of VLDL TG formation. Intakes of greater than 2 g n-3 fatty acid (equivalent to 6 g of most forms of fish oil) per day lower TG in a dose-dependent fashion. Up to 50% reduction in TG may be achieved with 15 g fish oil per day.
Changes in LDL-C and HDL-C are variable. Fish oil fatty acids have also been shown to inhibit platelet aggregation and improve cardiac arrhythmia in animal models. Dietary and pharmacological trials indicate that n-3 fatty acids reduce mortality from coronary heart disease. Fish oils appear to be safe and well tolerated.
Management of hyperlipidaemia in the elderly
• Prevalence of atherosclerotic cardiovascular disease: greatest in old age.• Associated cardiovascular risk: lipid levels become less predictive, as do other risk factors apart from age itself.
• Benefit of statin therapy: maintained up to the age of 80 years but evidence is lacking beyond this.
• Life expectancy and statin therapy: lives saved by intervention are associated with shorter life expectancy than in younger patients, and so the impact of statins on quality-adjusted life years is smaller in old age.
Dyslipidaemia in pregnancy
Lipid metabolism: lipid and lipoprotein levels increase during pregnancy. This includes an increase in LDL-C which resolves post-partum. Remnant dyslipidaemia and hypertriglyceridaemia may be exacerbated during pregnancy.Treatment: dyslipidaemia is rarely thought to warrant urgent treatment so pharmacological therapy is usually contraindicated when conception or pregnancy is anticipated. Teratogenicity has been reported with systemically absorbed agents, and non-absorbed agents may interfere with nutrient bioavailability.
Monitoring: cardiovascular disease is very unlikely amongst women of child-bearing age, but is possible in women with severe risk factor profiles or familial hypercholesterolaemia, when pre-conception cardiovascular review can be considered to ensure that the patient will be able to withstand the demands of pregnancy and labour.
Patients with predominant hypertriglyceridaemia who do not respond to lifestyle intervention can be treated with fibrates, fish oil or nicotinic acid, depending on individual response and tolerance.
If target levels are not achieved, the fibrates or nicotinic acid and fish oil can be combined. Massive hypertriglyceridaemia may require more aggressive limitation of dietary fat intake (< 10-20% energy as fat). Any degree of insulin deficiency should be corrected because insulin is required for optimal activity of lipoprotein lipase. The initial target for patients with massive hypertriglyceridaemia is TG < 10 mmol/L (880 mg/dL), to reduce the risk of acute pancreatitis.
Mixed hyperlipidaemia can be difficult to treat. Statins alone are less effective first-line therapy once fasting TG exceeds around 4 mmol/L (350 mg/dL). Fibrates alone are first-line therapy for dysbetalipoproteinaemia, but they may not control the cholesterol component in other forms of mixed hyperlipidaemia. Combination therapy is often required.
Mixed hyperlipidaemia
Effective combinations include: statin plus fish oil when TG is not too high; fibrate plus ezetimibe; statin plus nicotinic acid; or statin plus fibrate. Fibrates are effective in combination with statins but the risk of myopathy is increased. There is some evidence that fenofibrate is safer than gemfibrozil in this regard.
The effect of drug therapy can be assessed after 6 weeks (12 weeks for fibrates), and it is prudent to review side-effects, lipid response, CK and liver function tests at this stage. Follow-up should encourage continued compliance (especially diet and exercise), and include monitoring for side-effects and cardiovascular symptoms or signs, and measurement of weight, blood pressure and lipids, as well as review of absolute cardiovascular disease risk status.
Monitoring of therapy
Further assessment of CK and liver function tests is required only if relevant symptoms occur, or if statins are used in combination with fibrates, nicotinic acid or other drugs that may interfere with their clearance. If myalgia or weakness is associated with CK elevation > 5-10 times the upper limit of normal, or if sustained alanine aminotransferase (ALT) elevation > 2-3 times the upper limit of normal (and not accounted for by fatty liver) is detected, treatment should be interrupted and alternative therapy sought.
In combined therapy,
fibrates should be given in the morningstatins at night so that the peak dosages do not overlap.
• Lowering of LDL cholesterol by reducing saturated fat intake.• Lowering of Triglyceride levels by reducing consumption of sugary and processed foods.
• Reduction of Homocysteine levels by supplementation with Vitamins B6 and B12, and folic acid.
• Increased antioxidant activity by higher consumption of fruits and vegetables.
• Lowering of fibrinogen and growth factors by cutting back on foods such as red meat, dairy products, poultry and eggs.
Dietary fats and their food sources
• Raises LDL ("bad" cholesterol)• Little effect on HDL ("good" cholesterol) or triglycerides
• Fatty meats (beef, pork)
• Poultry skin
• Butterfat (in whole milk, cream, ice cream, cheese)
• Tropical oils (coconut, palm)
• Chocolate
• Saturated fat
• Lowers LDL if substituted for saturated fat
• Keeps HDL up
• Olive oil
• Peanut oil
• Canola oil
• Monounsaturated fat
• Linoleic acid in moderation can lower LDL
• Sunflower oil
• Sesame oil
• Corn oil
• Soybean oil
• Polyunsaturated fat
• Lowers triglycerides
• "Thins" the blood
• All fish, especially fatty fish, such as salmon and mackerel
• Plant sources, such as walnuts, canola, and flaxseed oils
• Omega-3 fats
• Raises LDL
• Little effect on HDL but at high levels can lower HDL
• Hydrogenated fats, margarine, vegetable shortening, nondairy creamer and whipped toppings Snack foods (potato chips, cookies, cakes)
• Peanut butter that contains hydrogenated fat (except all-natural varieties)
• Trans fatty acids
Foods to avoid for high cholesterol
Avoid saturated fat and oils, such as butter, bacon drippings, palm oil, and coconut oil. Replace these with soft tub margarine or vegetable oils, such as olive, safflower, soy, corn, canola, or peanut oil.
Limit trans fatty acids or partially hydrogenated vegetable oils, such as those found in hard margarines, snack crackers, cookies, chips, Hydrogenation is a process that makes the fat solid or semisolid.
Limit fatty meats such as corned beef, pastrami, ribs, steak, ground meat, sausage,. Limit high-cholesterol organ meats (liver and kidney) and egg yolks.
Replace with skinless chicken or turkey, lamb, fish, and meatless main dishes including beans, peas, pasta, or rice.
Limit servings of meat, poultry, and fish to 2 oz (56.7 g) to 3 oz (85.1 g) (a serving is about the size of a deck of playing cards), twice a day, or no more than 5 oz (141.8 g) per day. Limit milk products that contain more than 1% milk fat, such as cream, most cheeses, and nondairy coffee creamers or whipped topping (which often contain coconut or palm oils). Replace with skim or low-fat milk (0% to 1% fat) and low-fat cheeses that contain fewer than 3 grams of fat per ounce. Avoid fast foods (such as hamburgers, fried chicken, and tacos),
Eggs: Are they good or bad for my cholesterol?
Eggs are high in cholesterol, and a diet high in cholesterol can contribute to elevated blood cholesterol levels. But the extent to which dietary cholesterol raises blood cholesterol levels isn't clear. Many scientists believe that saturated fats and trans fats have a greater impact than dietary cholesterol in raising blood cholesterolIf you are healthy, it is recommended that you limit your dietary cholesterol intake to less than 300 milligrams (mg) a day.
If you have cardiovascular disease, diabetes or high LDL cholesterol, limit dietary cholesterol intake to less than 200 mg a day.
The yolk of one large egg has about 213 mg of cholesterol. If you eat an egg on a given day, it may be a good idea to limit or avoid other sources of cholesterol for the rest of that day.
Very low-fat diets
Although very low-fat diets may indeed lower cholesterol levels, they are not recommended. Very low-fat diets usually allow less than 15% of total calories from fat.In comparison, a cholesterol-reducing diet allows 25% to 35% of calories to come from total fat, with 7% from saturated fat.
A diet with less than 25% of its calories from fat can increase triglycerides and decrease HDL (good) cholesterol. Such a diet may deplete your body of other important nutrients and vitamins.
lifestyle changes
Follow the Therapeutic Lifestyle Changes (TLC) diet.The goal is to reduce the amount of saturated fat you eat. The TLC diet helps you learn to make better food choices by picking lean meats, low-fat or nonfat products, and good fats like olive and canola oils.
Lose weight,Be more active.
Quit smoking
How is it treated?
Omega 3 Fatty Acids (Fish Oils)
N-3 polyunsaturated fatty acids (n-3 PUFAs) are present in high concentration in fish and in flax seeds. The most widely used n-3 PUFAs for the treatment of hyperlipidemias are the two active molecules in fish oil: eicosapentanoic acid (EPA) and decohexanoic acid (DHA). N-3 PUFAs have been concentrated into tablets and decrease fasting triglycerides in doses of 3–4 g/d. Fish oils can result in an increase in plasma LDL-C levels in some patients.Fish oil supplements can be used in combination with fibrates, niacin, or statins to treat hypertriglyceridemia. In general, fish oils are well tolerated and appear to be safe, at least at doses up to 3–4 g. Although fish oil administration is associated with a prolongation in the bleeding time, no increase in bleeding has been seen in clinical trials. A lower dose of omega 3 (about 1 g) has been associated with reduction in cardiovascular events in CHD patients and is used by some clinicians for this purpose.
The NCEP ATPIII guidelines published in 2001 recommend that all adults over age 20 should have plasma levels of cholesterol, triglyceride, LDL-C, and HDL-C measured after a 12-hour overnight fast. In most clinical laboratories, the total cholesterol and triglycerides in the plasma are measured enzymatically, and then the cholesterol in the supernatant is measured after precipitation of apoB-containing lipoproteins to determine the HDL-C. The LDL-C is estimated using the following equation:
LDL-C = total cholesterol – (triglycerides/5) – HDL-C.
(The VLDL-C is estimated by dividing the plasma triglyceride by 5, reflecting the ratio of cholesterol to triglyceride in VLDL particles.) This formula is reasonably accurate if test results are obtained on fasting plasma and if the triglyceride level does not exceed ~300 mg/dL; by convention it cannot be used if the TGs are >400 mg/dL. The accurate determination of LDL-C levels in patients with triglyceride levels >300 mg/dL requires application of ultracentrifugation techniques or other direct assays for LDL-C. Further evaluation and treatment is based primarily on the plasma LDL-C level and the assessment of overall cardiovascular risk.The VLDL-C is estimated by dividing the plasma triglyceride by 5, reflecting the ratio of cholesterol to triglyceride in VLDL particles.
What are xanthomas?
Xanthomas are skin lesions caused by the accumulation of fat in macrophage s in the skin and more rarely in the layer of fat under the skin.
Xanthomas are classified into the following types based on where they are found on the body and how they develop.
eruptive and tuberous xanthomas may disappear after systemic treatment,
while other such as tendinous xanthomas, will not. least specific sign for hyperlipidemia is xanthelasma, because at least 50% of the patients with this finding have normal lipid profiles.


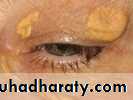
xanthelasma
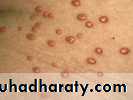
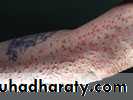
Lesions typically erupt as crops of small, red-yellow papules
Most commonly arise over the buttocks, shoulders, arms and legs but may occur all over the body
Rarely the face and inside of the mouth may be affected
Lesions may be tender and usually itchy
Lesions may resolve spontaneously over a few weeks
Associated with hypertriglyceridaemia
Eruptive xanthomas
Plane xanthomas
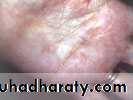
(Palmar xanthoma)Lesions are flat papules or patches that can occur anywhere on the body
Lesions on the creases of the palms are indicative of a specific pattern of increased lipids in blood called type III and type IIA dysbetalipoproteinaemia
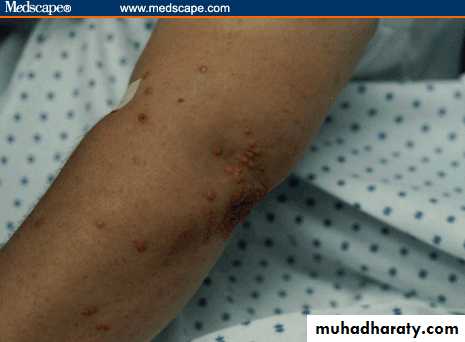
Eruptive xanthomas in a patient with diabetes and poor glycemic control.
Eruptive Xanthomaspainless, yellowish papules on an erythematous base that present as grouped lesions on the torso, especially the elbows ,chest, and buttock regions. Triglyceride levels are very high, often exceeding 2,000 mg/dL.They may appear in a patient with diabetes who has poor glycemic control.
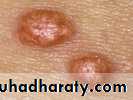
Tuberous and Tuberoeruptive Xanthomas
firm and nontender cutaneous and subcutaneous nodules that may be confused with gouty tophi. They occur on extensor surfaces of the large joints or hand ,knees, buttocks, and elbow ,heels . as well as in areas of prior trauma. pressure areas .Tuberous xanthomas are circular, raised lesions that display a yellowish-orange hue,associated with hypertriglyceridemia, but they are also seen in patients with hypercholesterolemia .
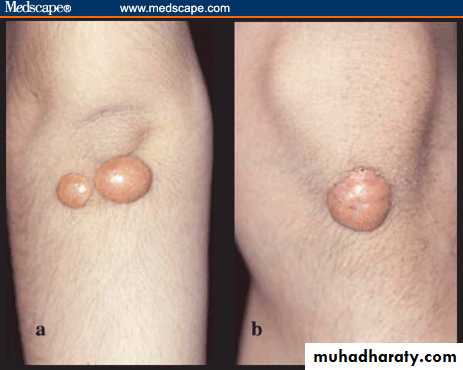
Examples of tuberous xanthomas on the (a) back of the elbow and (b) front of the knee.
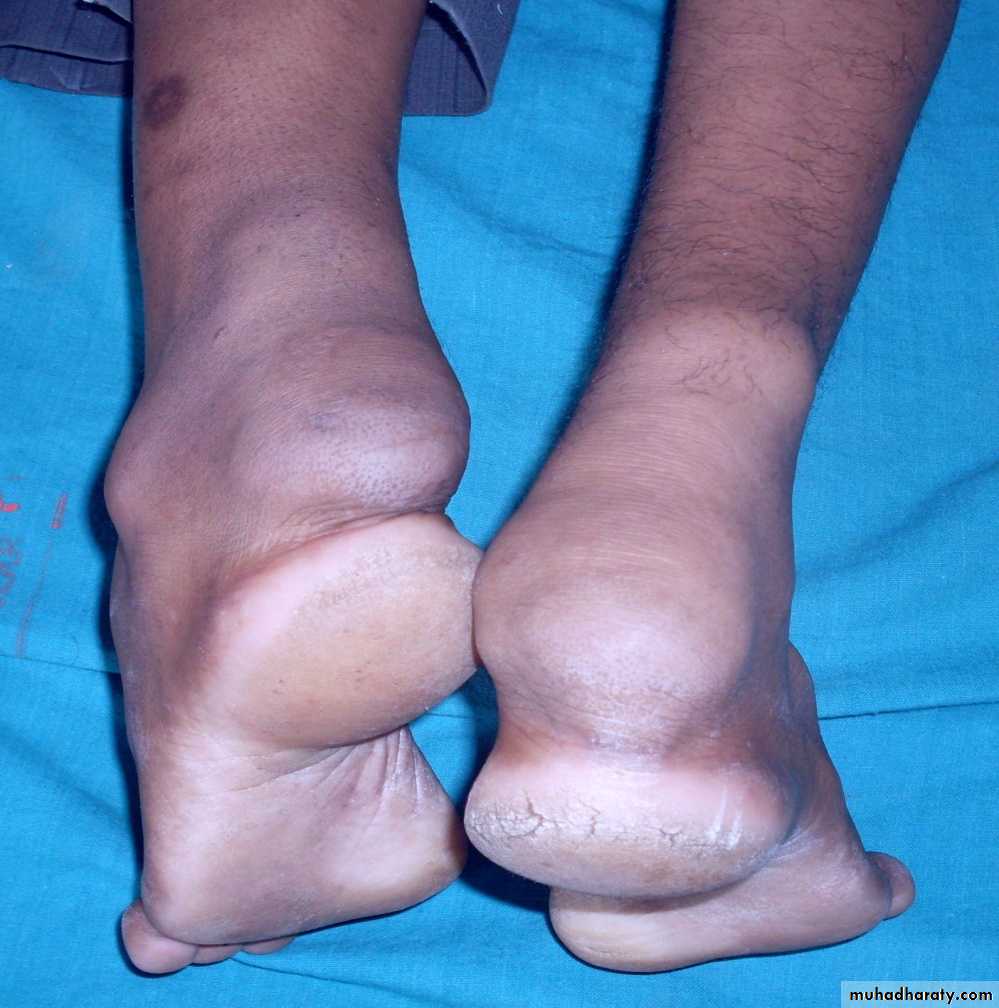
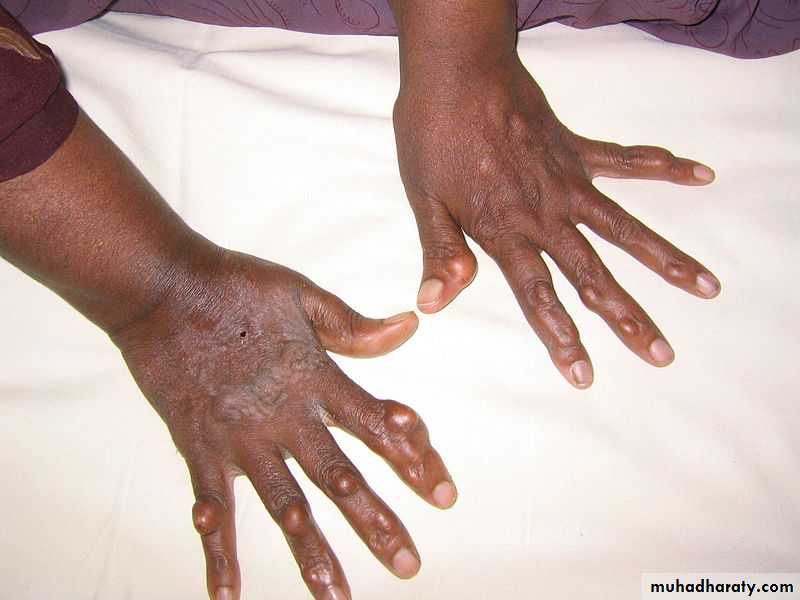
Tendinous xanthomacharacterized by papules and nodules ,associated with Type II hyperlipidaemia . Most commonly found in the tendons of the hands, feet, and Achilles tendon
Hypertriglyceridaemia is a recognised cause of acute pancreatitis, being responsible for between 1% and 4% of cases. In severe hypertriglyceridaemia, large lipoproteins including chylomicrons impair circulation in the capillary beds. This can restrict blood flow in the pancreas, which results in ischaemia of the acinar structures,, pancreatic enzymes escape from the acinar cells and initiate hydrolysis of triglycerides to free fatty acids. Localised concentrations of unbound free fatty acids are proinflammatory and cause further damage to the acinar cells and local vasculature. More pancreatic enzymes are released and the destructive cycle continues to eventually produce acute pancreatitis .
Hypertriglyceridaemia interferes with the serum amylase assay and can produce a falsely low result—serum amylase can seem to be within normal limits despite clinical and radiological evidence of acute pancreatitis.
Urine amylase to creatinine ratio can be measured to diagnose acute pancreatitis, and the result of this test is less likely to be affected by hypertriglyceridaemia. Alternatives are the removal of lipids before serum amylase measurement by using ultracentrifugation..
Myocardial infarction and other types of tissue injury generate changes in plasma proteins known as the acute phase response.
Maximal postinfarction reductions in total cholesterol occur at days 4 to 5 with levels 47% below baseline; low and high density lipoprotein cholesterol fractions decrease to their nadir on day 7 to concentrations that are 48% and 32% below baseline, respectively.
Triglyceride levels increase after acute myocardial infarction to a maximal level that is 58% above baseline on day 7. These alterations in lipid and lipoprotein levels generally stabilize by 2 months after the acute event.
In patients with acute myocardial infarctions cholesterol levels are no longer valid after 24 h from presentation because acute MI causes a rapid decline in serum levels of total cholesterol, LDL cholesterol, and HDLcholesterol. Nevertheless, several large-scale epidemiologic studies have shown that the total /HDL and the LDL /HDL cholesterol ratio are also strong predictors of coronary events. ratios remained unchanged. Therefore, most experts recommend
measuring the serum cholesterol levels within the
first 24 h after the onset of an acute MI or measure the cholesterol ratios.
The ratios of total cholesterol to HDL cholesterol and LDL cholesterol to HDL cholesterol that have been reported to correlate with the development of acute coronary events are 4.5
(Ideally, one should strive for ratios of 2 or 3 (less than 4)).and 2.5, respectively.
EXOGENOUS LIPID METABOLISM
Animal products, composed of substantial amounts of cholesterol and triglycerides, are eaten regularly by most people. Dietary fats are broken down in the gut to free cholesterol and fatty acids, which are transported across cell membranes into the enterocyte. Here, they are re-esterified into cholesteryl ester and triglycerides and then packaged onto apo B48. These particles gain access to the plasma through the thoracic duct and acquire other apolipoproteins in part by transfer from HDL, and these mature chylomicrons circulate to peripheral tissues.LPL, bound to the capillary endothelium in tissues such as adipose tissue and muscle, is activated by apo CII on chylomicrons, and fatty acids hydrolyzed from triglycerides by LPL are released and transported into adipose tissue for storage or muscle for energy. This process also requires apo AV, an apolipoprotein transported in HDL that appears to facilitate the interaction between LPL and triglyceride-rich lipoproteins.
Progressive hydrolysis of triglyceride converts chylomicrons into chylomicron remnants, which are relatively enriched in cholesteryl esters. Chylomicron remnants are removed in the liver by species that bind apo E: LRP, the LDL receptor, and cell surface glycosaminoglycans. Chylomicrons are large, and it is unlikely that they contribute to atherosclerosis.
Chylomicron remnants are enriched in cholesteryl esters, the major lipid component of the atherosclerotic lesion, and small enough to enter the subendothelial space, where they are taken up by macrophages. Remnants are atherogenic, consistent with the idea that the progression of atherosclerotic lesions can occur in the postprandial state, a potential contributor to morbidity not assessed by current practices that focus on the measurement of fasting lipoproteins.
Chylomicrons are not soluble. Their presence causes the “tomato soup” appearance of blood drawn after a fatty meal. Because they are mostly triglycerides, they float to the top of serum that is refrigerated overnight, leaving a layer of cream on the top of the sample. The detection of chylomicrons in fasting serum has clinical relevance because it indicates a risk for pancreatitis and other elements of the chylomicronemia syndrome.
ENDOGENOUS LIPID METABOLISM
Fats deposited in the liver are further metabolized into component lipid species, re-esterified as cholesteryl ester and triglycerides, and either stored in hepatocytes or exported as lipoproteins ( Fig. 217-1 ). The liver produces the triglyceride-rich lipoprotein VLDL. Its rate of production appears to depend on the availability of triglycerides. Apo B100 is the major apolipoprotein of VLDL, but under normal conditions the regulation of the apo B gene does not appear to play an appreciable role in the control of VLDL synthesis.Apo B transcription appears to be mostly constitutive, and production of the apo B100 protein depends on its cotranslational stabilization. As the message is translated into protein, the presence of triglyceride stabilizes the peptide and allows continued addition of amino acids. In the absence of triglycerides, the apo B molecule is degraded.
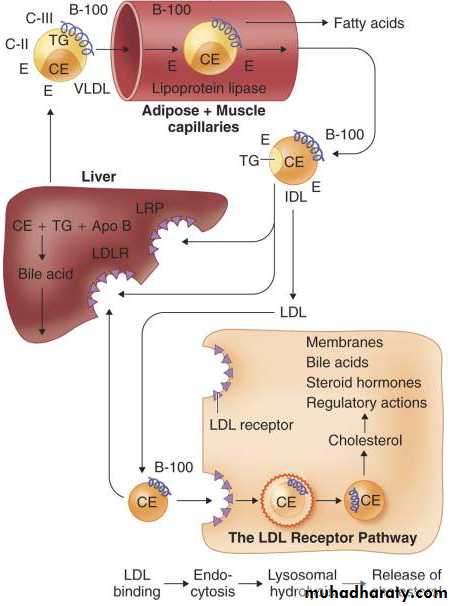
FIGURE 217-1 Endogenous lipid metabolism. In the liver, triglycerides (TGs), cholesteryl esters (CEs), and apolipoprotein B100 are packaged as (VLDL) particles. TG is hydrolyzed by lipoprotein lipase (LPL) to generate (IDL), which is further metabolized to generate (LDL). This particle can be removed by the liver or by peripheral cells. Cholesterol derived from LDL regulates several processes and can be used for the synthesis of bile acids, steroid hormones, and cell membranes.
The transfer of triglycerides to the growing apo B peptide is mediated by microsomal transfer protein (MTP). Mutations in MTP cause abetalipoproteinemia, a rare disease characterized by the absence of circulating apo B. In the absence of apo B, the metabolism of fat-soluble vitamins (normally carried in lipoproteins) is disrupted and patients suffer from multisystem defects including severe neurologic dysfunction and retinopathy presumably caused by deficiency of vitamins E and A. Drugs that interfere with MTP function lower lipids but, not surprisingly, cause the accumulation of triglyceride in the liver.
The apo B gene is normal in patients with abetalipoproteinemia. Mutations in the apo B gene cause another condition known as hypobetalipoproteinemia, caused by shortened forms of the apo B protein. Subjects with hypobetalipoproteinemia have very low, but not absent, levels of circulating lipids and appear to be healthy.
Nascent VLDL containing one apo B100 molecule per particle is secreted into the plasma, where it acquires apo E, apo CII, and apo CIII. In a process analogous to that occurring with chylomicrons, apo CII on VLDL activates LPL, and fatty acids hydrolyzed from triglycerides by LPL are released in capillary beds and transported into tissues.
With continued hydrolysis and the loss of both phospholipids and apolipoproteins to HDL, VLDL is converted to IDL, a cholesteryl ester–rich particle with an apolipoprotein complement of only apo B and apo E. These particles, like chylomicron remnants, are thought to have high atherogenic potential. Unlike chylomicron remnants, IDLs are included in current management schemes because reporting of LDL cholesterol levels by most clinical laboratories includes IDL.
IDL can be taken up by either the LRP or the LDL receptor in the liver. In the presence of a normal apo E molecule, IDLs are converted to LDL, with one molecule of apo B100 per particle and cholesteryl ester with essentially no triglycerides. About 75% of LDL is removed from the plasma by the LDL receptor pathway, and most of this takes place in the liver.
The uptake of LDL results in the migration of LDL particles to lysosomes, where cholesterol is released for (depending on the cell type) plasma membrane localization, bile acid synthesis, steroid hormone synthesis, and interaction with Scap for the control of SREBP activation. A minority of LDL enters the subendothelial space of the vascular wall, where its modification by oxidation or other processes promotes its uptake by macrophages in atherosclerotic lesions.
Most VLDL particles are large and not thought to promote vascular disease. However, some small VLDL particles as well as IDL and LDL are atherogenic. Because the majority of VLDL is triglyceride, most patients with elevated levels of fasting triglyceride have either increased numbers of VLDL particles or increased content of triglycerides in each VLDL. Unlike most other lipoproteins, LDL has a fairly long residence time with a plasma half-life of 2 to 5 days.
Accordingly, the detection of elevated levels of fasting cholesterol usually reflects the presence of either increased numbers of LDL particles or increased cholesteryl ester in each LDL. LDL also exists in a range of sizes. Small dense LDL tends to occur in the setting of concomitant hypertriglyceridemia. This type of lipoprotein is thought to have greater atherogenic potential than larger LDL species, perhaps because of more facile access to the vascular wall and greater susceptibility to oxidative modification.
Lipoprotein particle size and number can be quantified by nuclear magnetic resonance techniques, but it is not clear that these data provide diagnostic advantages beyond the determination of total cholesterol, triglycerides, LDL cholesterol, and HDL cholesterol.
REVERSE CHOLESTEROL TRANSPORT AND HIGH-DENSITY LIPOPROTEIN METABOLISM
Lipid metabolism is extremely dynamic. At the same time lipoproteins are processed to modify their nonpolar lipids, particles are interacting with each other, exchanging surface materials, apolipoproteins, and nonpolar lipids. HDL is an important reservoir for components cast off during the metabolism of other lipoproteins as well as lipids discarded by cells.Nascent HDL is generated by the liver and intestine as a phospholipid disc containing apo AI and apo AII. It accepts unesterified (free) cholesterol (UC in Fig. 217-2 ) and phospholipids shed from cells. This unesterified cholesterol is converted to cholesteryl ester by the action of LCAT and stored in the center of the disc, allowing it to become a spherical particle. The particle is further modified as a consequence of the action of LPL on triglycerides in apo B–containing lipoproteins.
As the core triglycerides of VLDL are metabolized, the particle collapses, leaving redundant surface lipids (phospholipid in the form of lecithin and unesterified cholesterol) and excess apolipoproteins such as apo CII, apo CIII, and apo E that are transferred to HDL. LCAT again esterifies the cholesterol to increase the content of cholesteryl ester in the HDL.
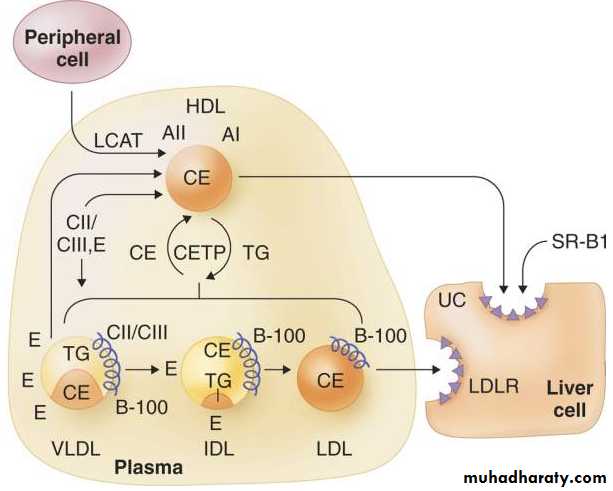
FIGURE 217-2 Reverse cholesterol transport and HDL metabolism. Unesterified cholesterol (UC) in peripheral cells can be transferred to HDL and esterified by lecithin-cholesterol acyltransferase (LCAT). This cholesterol ester (CE) in HDL can be transferred to the liver directly through scavenger receptor B1 (SR-B1). Alternatively, it may be transferred to apolipoprotein B100–containing lipoproteins in exchange for triglycerides through the action of cholesteryl ester transfer protein (CETP).
Reverse cholesterol transport is the beneficial process by which cholesterol present in peripheral cells such as foam cells in a growing atherosclerotic lesion is transported back to the liver for excretion. There are at least two well-defined pathways mediating this transfer. First, after accepting cholesterol from peripheral cells and esterifying it through the action of LCAT, HDL can interact directly with the liver by binding to SR-B1 and transferring cholesteryl ester to the hepatocyte.
Second, HDL can transfer cholesteryl ester to apo B100–containing lipoproteins such as VLDL (bottom of Fig. 217-2 ) through the action of CETP. This cholesteryl ester can ultimately be transported to the liver after conversion of VLDL to IDL to LDL and uptake by the LDL receptor (LDLR in Fig. 217-2 ). This pathway is not direct because the transfer of cholesteryl ester to apo B–containing lipoproteins results in cholesterol-enriched particles that may be taken up by foam cells in atherosclerotic plaques before being cleared by the liver.
Humans with genetic defects in CETP have high HDL levels and appear to be healthy. An inhibitor of CEPT, torcetrapib increases HDL cholesterol but increases adverse events, perhaps in part related to its adverse effects on blood pressure.
Nuclear receptors are transcription factors that are activated by ligand binding to increase the expression of specific sets of genes. Several nuclear receptors are activated by lipids, play important roles in systemic lipid metabolism, and are current or potential targets of medications for altering lipids in patients.
LIPID-ACTIVATED NUCLEAR RECEPTORS AND LIPID METABOLISM
Peroxisome proliferator–activated receptors (PPARs, so named because their activation increases peroxisomes in rodents) are thought to be activated by fatty acids. There are at least three types, PPARa, PPAR?, and PPARd. PPARa appears to be particularly important in liver and muscle. It stimulates the expression of genes mediating fatty acid oxidation as well as those promoting the formation of HDL.
Fibrate drugs such as gemfibrozil and fenofibrate work by activating PPARa. They lower triglycerides, by accelerating the oxidation of fatty acids in the liver so that less lipid is available for stabilizing apo B100 in VLDL secretion, and elevate HDL, by increasing expression of apo AI, LPL, and other genes. PPAR? is expressed mostly in adipose tissue and macrophages but also has measurable effects in other tissues such as muscle.
It increases the expression of genes promoting the development of fat tissue and appears to suppress certain mediators of chronic inflammation. Thiazolidinedione drugs such as pioglitazone and rosiglitazone work by activating PPAR?. They lower blood glucose in people with diabetes by decreasing insulin resistance, a complex process that also results in multiple lipid effects including lower triglycerides and higher HDL (expected to be beneficial) and higher LDL (expected to be detrimental).
The overall effects appear to be favorable. Combined PPARa-PPAR? agonists are in development. PPARd is expressed widely, has multiple potential effects that include the regulation of fatty acid oxidation, and is the subject of intense pharmacologic investigation.
Two other nuclear receptors are activated by lipids and modulate lipid physiology. Liver X receptors (LXRa and LXRß) are activated by oxidized derivatives of cholesterol. In the liver, they promote the synthesis of fatty acids and triglycerides in addition to stimulating both the conversion of cholesterol into bile acids and the excretion of bile acids into the gut. In the intestine, they suppress the absorption of cholesterol. The farnesoid X receptor (FXR) is activated by bile acids. FXR stimulates the secretion of bile acids into bile as well as the reabsorption of bile acids from the intestine.
Together, these receptors help orchestrate two important futile cycles in lipid metabolism. In one, fatty acids exported from the liver (in the form of triglycerides within VLDL) and from the intestine (in chylomicron triglycerides) are released to peripheral tissues by the action of LPL. Some are taken up by muscle, where their activation of PPARa accelerates their oxidation in mitochondria, yielding ATP.
Others enter adipose tissue, where they are re-esterified into triglycerides. From here, fatty acids are released in a process stimulated by catecholamines and glucagon. This process is complicated, involving hormone-sensitive lipase, a recently discovered enzyme known as adipose triglyceride lipase, and the remodeling of proteins that coat lipid droplets to alter their accessibility to lipases.
Following lipolysis, fatty acids bind to albumin and return to the liver, where they can fuel the production of more VLDL particles.
Nicotinic acid improves lipids in part by blocking the release of fatty acids from adipose tissue. A G protein–coupled receptor for nicotinic acid has been identified.
In another futile cycle, bile acids (formed from cholesterol and constituting the major pathway for excretion of cholesterol from the body) are secreted into the intestine through events stimulated by LXR and FXR. Bile acids are reabsorbed in the terminal ileum.
Treatment with bile acid sequestrants such as cholestyramine and colesevelam interrupts this enterohepatic circulation, and the increased excretion of cholesterol (in the form of bile acids) depletes cholesterol content in the liver, leading to the induction of LDL receptors resulting in lower circulating levels of LDL. This treatment also tends to elevate triglyceride levels, the result of de-repression of several processes mediated by FXR that decrease fatty acids and triglycerides.
The major lipids transported in the blood are triglycerides; between 70 and 150 g enter and leave the plasma daily compared with 1 to 2 g of cholesterol or phospholipid. Chylomicrons, the largest lipoproteins, carry exogenous triglyceride from the intestine via the thoracic duct to the venous system. In the capillaries of adipose and muscle tissue, 90% of chylomicron triglyceride is removed by a specific group of lipases.
Fatty acids and glycerol, derived from hydrolysis of chylomicrons, enter the adipocytes and muscle cells for energy use or storage. The liver then removes the remnant chylomicron particles. VLDL carries endogenous triglyceride primarily from the liver to the same peripheral sites (adipocytes and muscle cells) for storage or use.
The same lipases that act on chylomicrons quickly degrade endogenous triglyceride in VLDL, giving rise to intermediate density lipoproteins (IDL) that are shorn of much of their triglyceride and surface apoproteins. Within 2 to 6 h, this IDL is degraded further by removal of more triglyceride, giving rise to LDL, which in turn has a plasma half-life of 2 to 3 days. VLDL is, therefore, the main source of plasma LDL.
The fate of LDL is unclear: The liver removes about 70%, and active receptor sites have been found on the surfaces of hepatocytes and other cells that specifically bind to apolipoprotein B (apo B, the ligand associated with LDL that binds with LDL receptors) and remove most LDL from the circulation. A small but important amount of LDL appears to be removed from the circulation by non-LDL receptor pathways, including uptake by scavenger receptors on macrophages that may migrate into arterial walls, where they may become the foam cells of atherosclerotic plaques.
Hypercholesterolemia can result either from overproduction or defective clearance of VLDL or from increased conversion of VLDL to LDL. Overproduction of VLDL by the liver may be caused by obesity, diabetes mellitus, alcohol excess, nephrotic syndrome, or genetic disorders; each condition can result in increased LDL and TC levels and often is associated with hypertriglyceridemia.
Defective LDL clearance may be due to genetically determined structural defects in apo B (the ligand) that diminish the binding of apo B to otherwise normal LDL receptors. Alternatively, reduced clearance may be due to diminished numbers or abnormal function (low activity) of the LDL receptors, which may result from genetic or dietary causes. Genetically mediated abnormal LDL receptor function usually results from molecular defects in the protein structure of the receptors--the usual mechanism of the genetic disorders described below.
When dietary cholesterol (as a constituent of chylomicron remnants) reaches the liver, the resulting elevated levels of intracellular cholesterol (or a metabolite of cholesterol in the hepatocyte) suppress LDL-receptor synthesis; this suppression occurs at the level of transcription of the LDL gene. A reduced number of receptors results in higher levels of plasma LDL and therefore of TC. Saturated fatty acids also increase plasma LDL and TC levels; the mechanism of action is related to a reduced activity of LDL receptors.
Transport of Dietary Lipids (Exogenous Pathway)
The exogenous pathway of lipoprotein metabolism permits efficient transport of dietary lipids (Fig. 350-2). Dietary triglycerides are hydrolyzed by lipases within the intestinal lumen and emulsified with bile acids to form micelles. Dietary cholesterol, fatty acids, and fat-soluble vitamins are absorbed in the proximal small intestine. Cholesterol and retinol are esterified (by the addition of a fatty acid) in the enterocyte to form cholesteryl esters and retinyl esters, respectively. Longer-chain fatty acids (>12 carbons) are incorporated into triglycerides and packaged with apoB-48, cholesteryl esters, retinyl esters, phospholipids and cholesterol to form chylomicrons.Nascent chylomicrons are secreted into the intestinal lymph and delivered via the thoracic duct directly to the systemic circulation, where they are extensively processed by peripheral tissues before reaching the liver. The particles encounter lipoprotein lipase (LPL), which is anchored to proteoglycans that decorate the capillary endothelial surfaces of adipose tissue, heart and skeletal muscle (Fig. 350-2).
The triglycerides of chylomicrons are hydrolyzed by LPL, and free fatty acids are released. ApoC-II, which is transferred to circulating chylomicrons from HDL, acts as a cofactor for LPL in this reaction. The released free fatty acids are taken up by adjacent myocytes or adipocytes and either oxidized to generate energy or reesterified and stored as triglyceride. Some of the released free fatty acids bind albumin before entering cells and are transported to other tissues, especially the liver.
The chylomicron particle progressively shrinks in size as the hydrophobic core is hydrolyzed and the hydrophilic lipids (cholesterol and phospholipids) and apolipoproteins on the particle surface are transferred to HDL, creating chylomicron remnants. Chylomicron remnants are rapidly removed from the circulation by the liver through a process that requires apoE as a ligand for receptors in the liver. Consequently, few, if any, chylomicrons or chylomicron remnants are present in the blood after a 12-h fast, except in patients with disorders of chylomicron metabolism.
HDL Metabolism and Reverse Cholesterol Transport
All nucleated cells synthesize cholesterol, but only hepatocytes and enterocytes can effectively excrete cholesterol from the body, into either the bile or the gut lumen. In the liver, cholesterol is excreted into the bile, either directly or after conversion to bile acids. Cholesterol in peripheral cells is transported from the plasma membranes of peripheral cells to the liver and intestine by a process termed "reverse cholesterol transport" that is facilitated by HDL.
Nascent HDL particles are synthesized by the intestine and the liver. Newly secreted apoA-I rapidly acquires phospholipids and unesterified cholesterol from its site of synthesis (intestine or liver) via efflux promoted by the membrane protein ATP-binding cassette protein A1 (ABCA1). This process results in the formation of discoidal HDL particles, which then recruit additional unesterified cholesterol from the periphery. Within the HDL particle, the cholesterol is esterified by lecithin-cholesterol acyltransferase (LCAT), a plasma enzyme associated with HDL, and the more hydrophobic cholesteryl ester moves to the core of the HDL particle.
As HDL acquires more cholesteryl ester it becomes spherical, and additional apolipoproteins and lipids are transferred to the particles from the surfaces of chylomicrons and VLDLs during lipolysis.
HDL cholesterol is transported to hepatocytes by both an indirect and a direct pathway.
HDL cholesteryl esters can be transferred to apoB-containing lipoproteins in exchange for triglyceride by the cholesteryl ester transfer protein (CETP).
The cholesteryl esters are then removed from the circulation by LDL receptor–mediated endocytosis. HDL cholesterol can also be taken up directly by hepatocytes via the scavenger receptor class BI (SR-BI), a cell surface receptor that mediates the selective transfer of lipids to cells.
HDL particles undergo extensive remodeling within the plasma compartment by a variety of lipid transfer proteins and lipases. The phospholipid transfer protein has the net effect of transferring phospholipids from other lipoproteins to HDL. After CETP-mediated lipid exchange, the triglyceride-enriched HDL becomes a much better substrate for HL, which hydrolyzes the triglycerides and phospholipids to generate smaller HDL particles.
A related enzyme called endothelial lipase hydrolyzes HDL phospholipids, generating smaller HDL particles that are catabolized faster. Remodeling of HDL influences the metabolism, function, and plasma concentrations of HDL.

Don't drink your calories
Health tip
