
Neurology
د. حسين محمد جمعهاختصاصي الامراض الباطنة
البورد العربي
كلية طب الموصل
2011
Skeletal system metastases are the third most common metastases, after those of the pulmonary and hepatic systems.
Within the skeletal system, the spinal column is the most common site of metastases.
Metastatic cord compression is estimated to occur in 5-10% of patients with cancer (most commonly those with breast,prostate, and lung cancers) .
Most non-traumatic subarachnoid haemorrhage is caused by rupture of an intracranial aneurysm
Computed tomography is very sensitive in detecting acute subarachnoid haemorrhage but should not be relied on as the sole diagnostic investigation.
Lumbar puncture should be done at least 12 hours, after the onset of symptoms.
Computed tomography angiography is sensitive in detecting intracranial aneurysms, but catheter angiography is still the optimal investigation and may still be necessary .
Magnetic resonance imaging may be more sensitive than computed tomography in detecting subacute haemorrhage.
and is good for delayed presentations
Lumbar puncture
Lumbar puncture should be performed when computed tomography does not show blood but the physician suspects subarachnoid haemorrhage. Preferably 12 hours should have passed since the onset of symptoms before performing lumbarpuncture. Some evidence exists that lumbar puncture is most useful if clinical presentation is a few days after the haemorrhage.
Four tubes of cerebrospinal fluid should be
collected, and the first and fourth examined fornon-diminishingly raised levels of red blood cells. By using spectrophotometry, a bloody tap can be distinguished from true
subarachnoid bleeding by the levels of bilirubin and oxyhaemoglobin in the cerebrospinal fluid. These levels should still be detectable up to two weeks after onset of symptoms.
Intravenous Thrombolytic Therapy
for Acute Ischemic Stroke87% of all strokes worldwide are ischemic in origin (caused by in situ thrombosis, embolism, or systemic hypoperfusion). In 2007, stroke accounted for 1 of every 18 deaths in the United States. According to one report, the 30-day mortality for ischemic stroke was 8 to 12% for
nejm.org june 2, 2011
people 45 to 64 years of age.
In the Framingham Heart Study, among survivors of an ischemic stroke who were 65 years of age or older and were evaluated 6 months after the event,
50% had some evidence of hemiparesis,
30% were unable to walk without assistance,
19% had aphasia, and 26% were institutionalized.
The estimated direct medical cost of stroke in the United States was $25 billion in 2007.
In 1996, the Food and Drug Administration (FDA)
approved the use of intravenous rt-PA for thetreatment of acute ischemic stroke after the National Institute of Neurological Disorders and
Stroke Recombinant Tissue Plasminogen Activator (NINDS rt-PA) Stroke Study was completed.
In part 1 of this study, 291 patients with acute
ischemic stroke were randomly assigned, within3 hours after the onset of the stroke, to either
intravenous rt-PA or placebo. The primary end
point was the rate at 24 hours of either complete
neurologic recovery or neurologic improvement, as
indicated by an improvement of at least 4 points
above baseline values on the National Institutes
of Health Stroke Scale (NIHSS) (a 42-point scale
that quantifies neurologic deficits in 11 categories,
with higher scores indicating more severe deficits).
In this part of the trial, no significant difference
was seen in the primary end point between patients receiving rt-PA and those receiving placebo (51% and 46%, respectively; relative risk with rt-PA, 1.1; 95% confidence interval [CI], 0.8 to 1.6;P = 0.56).
Intravenous administration of t-PA within 3 hours
after the onset of stroke increases the probabilityof a favorable outcome. Recommended protocols
for selecting patients for treatment with intravenous rt-PA are adapted from the inclusion
and exclusion criteria from the NINDS rt-PA trial
(Table 1). On the basis of results of ECASS III,17
some stroke centers now treat patients who present from 3 to 4.5 hours after stroke onset; however,at present, the FDA has approved only rt-PA treatment delivered within 3 hours after stroke
onset.
The timing of the onset of stroke should be
determined with as much certainty as possible byobtaining first-hand information. If the onset was
not observed, the time when the patient was last
seen to be neurologically normal should be considered the time of stroke onset. Although this
recommendation may exclude some eligible patients,it ensures that those whose stroke occurred outside the time limit for a favorable risk-to-benefit ratio will not be treated.
A rapid examination with the use of the NIHSS
will help to quantify the neurologic deficit. Manyprotocols exclude patients who have mild deficits,
since their prognosis for recovery is good without
thrombolytic therapy.19,20 However, treatment
should be initiated on the basis of the assessment
of a disabling deficit rather than on a defined
lower limit for the NIHSS score. For example, isolated aphasia or hemianopia is a disabling deficit despite an NIHSS score of 2 or 3.
Rapidly resolving deficits may complicate decision
making. If the residual deficit continues to
be disabling, treatment should be undertaken despite the improvement. Occasionally, rapid recovery is later followed by clinical worsening.
Patients should therefore be observed closely and
reevaluated frequently during the first 24 hours
after the onset of stroke.
Another common concern regarding eligibility
for intravenous thrombolysis is poorly controlledblood pressure. In patients receiving intravenous
rt-PA, markedly elevated blood pressure
may increase the risk of hemorrhage. Current
guidelines recommend treatment to achieve
a systolic blood pressure of 185 mm Hg or lowerand a diastolic blood pressure of 110 mm Hg or
lower before intravenous rt-PA is administered.
One or two doses of labetalol may be used to
bring blood pressure below these limits, but ifthe response is not rapid, treatment with intravenous nicardipine or occasionally sodium nitroprusside may be started, with the dose rapidly adjusted to achieve blood-pressure control.
A CT scan of the brain should be obtained
before the start of treatment and examined forhemorrhage or early ischemic changes. If a focal
area of low density is seen that involves more
than one third of the middle-cerebral-artery territory,most treatment protocols recommend withholding thrombolytic therapy, because in some
studies this finding (which suggests irreversible
injury) has been predictive of subsequent hemorrhagic transformation of the infarct.
Laboratory
studies that should be obtained before
the initiation of thrombolytic therapy include,
at a minimum, a platelet count, measurement of
glucose levels, and assessment of the prothrombin
time. The platelet count should be greater than
100,000 per cubic millimeter, the prothrombin
time less than 15 seconds (or the INR <1.7), and
the glucose level greater than 50 mg per deciliter
(2.7 mmol per liter) before rt-PA is administered.
The patient and family members must be informed
of the benefits and risks of intravenousrt-PA therapy before it is initiated. Specifically, they
should be told that the benefits include an absolute increase in the odds of a good outcome of
11 to 13 percentage points and a 6% risk of intracerebral hemorrhage possibly causing neurologic worsening or death. Some hospitals choose to use a formal consent form, but at a minimum, the consent process should be documented in the medical record.
The FDA-approved dose of intravenous rt-PA is
0.9 mg per kilogram of body weight, with a maximum dose of 90 mg. A bolus of 10% of the doseis given over a period of 1 minute, with the remainder infused over a period of 60 minutes. Weight should be determined as reliably as is possible.
Reports of treatment with a lower dose of rt-PA
(0.6 mg per kilogram) in Japan suggest that it has
similar efficacy but the lower dose has not yet been
assessed in large, randomized trials.
Third-generation plasminogen activators, such
as tenecteplase and desmoteplase, are more fibrinspecific than rt-PA and cause less activation of
systemic lytic activity. These agents have been
tested in early-phase trials, with mixed results.
However, their clinical efficacy has not been established,
and neither agent should be used in patients
with acute ischemic stroke.
For the first 24 hours after treatment, patients
receiving rt-PA should be closely monitored in a
specialized stroke unit. If a stroke unit is not available,
admission to an intensive care unit is warranted
so that the patient can be evaluated frequently
by the nursing staff. Blood pressure shouldbe checked every 15 minutes for the first 2 hours,
every 30 minutes for the next 6 hours, and then
every hour for 16 hours. Antihypertensive therapy
with labetalol or, if necessary, intravenous nicardipine should be administered to maintain blood pressure at a level below 180 mm Hg systolic and 105 mm Hg diastolic.
Neurologic examination
with the use of the NIHSS should be performedevery 15 minutes for the first 2 hours,
every 30 minutes for the next 6 hours, and then every hour for 16 hours.
If a change in neurologicstatus is noted, the rt-PA infusion should be discontinued
and a CT scan obtained
No anticoagulant or antiplatelet therapy should be given for the first 24 hours after treatment with intravenous rt-PA. If a CT scan at 24 hours shows no evidenceof hemorrhage, antithrombotic therapy directed at secondary stroke prevention and tailored to the presumed cause of the stroke should be started.
In some stroke centers, a CT angiogram is obtained
after intravenous rt-PA has been administered
in order to examine the intracranial vasculature
for persistent arterial occlusions.
In patients with persistent arterial occlusion, one option is an intraarterial intervention: lytic therapy, mechanical clot disruption. Although this approach is not approved by the FDA in the treatment of acute
stroke, a randomized, controlled trial has suggested
a potential benefit from intraarterial lytic
therapy. However, intraarterial interventions
should be carried out only at experienced stroke
centers.
In one U.S. study, the cost of rt-PA was estimated
to be $2,750. In similar studies, the costswere £480 in the United Kingdom40 and $1,647
U.S. in Australia.41 Cost-effectiveness analyses in
general suggest that rt-PA therapy is more expensive than standard care for ischemic stroke in the short term, owing to the cost of the drug and the need for additional resources, but it is associated with lower costs in the long term, since it reduces the risk of subsequent disability.
Adverse Effects
The major complication of thrombolytic therapy for acute stroke is hemorrhage. Symptomatic intracranial hemorrhage occurs in 1.7 to 8.0% of treatedIn addition to age and NIHSS score,other independent risk factors for symptomatic intracranial hemorrhage include hypodensity on CT scanning, elevated serum glucose levels,and persistence of proximal arterial occlusion for more than 2 hours after administration of the rt-PA bolus. Hemorrhagic transformation of
ischemic infarcts without clinical change (asymptomatic hemorrhage) occurs more frequently
than symptomatic hemorrhage and may be
associated with reperfusion and, in some cases,
clinical improvement.
Serious systemic (extracranial) hemorrhage has been reported in 0.4 to 1.5% of patients. Recommendations for the treatment of intracranial or serious systemic bleeding after thrombolytic therapy often include the administration of cryoprecipitate and platelets,although evidence-based guidelines for such an approach are lacking.
Angioedema of the tongue, lips, face, or neck
occurs in 1 to 5% of patients receiving intravenousrt-PA.54,55 In most cases, the symptoms are
mild and resolve rapidly. Concomitant use of angiotensin-converting–enzyme inhibitors is strongly associated with this complication. Treatment includes glucocorticoids and antihistamines. In rare cases, edema of the pharynx is sufficiently severe to compromise breathing, and intubation may be necessary.
Areas of Uncertainy
More than half the patients with ischemic strokewho are treated with intravenous rt-PA do not
have complete or near-complete recovery (defined
as a score of 0 or 1 on the modified Rankin
scale). Lack of recovery may reflect an absence
of reperfusion of the occluded artery or reperfusion
that occurs too late to restore function.
Advanced imaging techniques that involve multimodal MRI or CT have the potential to
distinguish reversible ischemic injury from irreversible infarction and thus to identify patients
who are likely to benefit from thrombolytic therapy.
By identifying extensive areas of established
infarction, such imaging methods may
also help in selecting patients who are at high risk for intracranial hemorrhage and should therefore
not be treated with intravenous rt-PA
If a reliable pattern of reversibility can be identified, imaging might also be useful when the interval between the onset of stroke and presentation is prolonged or the time
of onset is not known.
Transcranial Doppler ultrasonography, which
has been used in some observational studies to
monitor the effect of lytic therapy, was shown
in these studies to be associated with a high rate
of recanalization of the occluded stroke-related
artery.
Transcranial ultrasonography was subsequently
evaluated in several small trials and wasshown to enhance the lytic effect of rt-PA,63-65 although some studies have suggested an increased
rate of hemorrhage with transcranial ultrasonography.
This approach, called sonothrombolysis,
has been implemented clinically as an adjunct to
rt-PA administration at some stroke centers.
Guidelines
Guidelines for the management of acute stroke issued by the American Heart Association (AHA) and
the European Stroke Organization recommend
treatment with intravenous rt-PA for patients who
meet the stated inclusion criteria, including presentation within 3 hours after the onset of stroke, and who do not meet any of the stated exclusion criteria.
Both groups have recently updated their guidelines to extend the treatment window to 4.5 hours.
The AHA Science Advisory and Coordinating
Committee also recommends that treatmentwithin the 3-hour to 4.5-hour time window be limited to patients who do not meet any of the ECASS III exclusion criteria. An American Academy of Emergency Medicine (AAEM) position statementadopted in 2002 concluded that intravenous rt-PA should not be considered the standard of care,citing the lack of data from trials confirming the NINDS study findings as well as concerns raised about the study.
treatment option when used in academic centers
and primary stroke centers. A policy statementapproved by the board of directors of the American College of Emergency Physicians in 2002 endorsed the use of intravenous rt-PA when it is administered according to the guidelines established by the NINDS study.
Summary
Stroke is the second leading cause of death worldwide,and the primary cause of serious, long-term disability in the United States. Joint guidelines from the American Heart Association (AHA) and American Stroke Association (ASA), as well as recent guidelines from the Eighth American College of Chest Physicians (ACCP)Combination Antiplatelet Agents for Secondary Prevention of Ischemic Stroke
Medscape 11/25/2008Conference on Antithrombotic and Antiplatelet Therapy, recommend aspirin, clopidogrel, or extended-release dipyridamole plus aspirin as acceptable firstline options for secondary prevention of ischemic events in patients with a history of ischemic stroke or transient ischemic attack (TIA).
The ACCP strongly recommends the combination of extended-release dipyridamole plus aspirin over aspirin monotherapy (highest level of evidence) and
suggests clopidogrel monotherapy over aspirin monotherapy (lower level of evidence).
The AHA-ASA guidelines suggest that either extended-release dipyridamole plus aspirin
or clopidogrel monotherapy should be used over aspirin monotherapy.Both guidelines recommend avoiding the combination of clopidogrel and aspirin
for most patients with previous stroke or TIA.The CHARISMA trial compared aspirin plus clopidogrel with aspirin alone in a population at high risk for atherothrombotic events using the composite outcome of myocardial infarction, stroke, and death from cardiovascular causes. Data from ESPRIT add to evidence that the combination of aspirin plus extended-release dipyridamole is superior to aspirin alone. The findings of the CHARISMA trial reinforce recommendations from both AHAASA and ACCP that the combination of aspirin and clopidogrel be reserved for special populations requiring this antiplatelet combination (e.g., those who have had coronary artery stenting).
Introduction
Nearly 800,000 people in the United States suffer a stroke each year, resulting in significant morbidity and mortality.[1] Roughly 90% of all strokes are ischemic in nature, with the remaining 10% resulting from intracerebral hemorrhage or subarachnoid hemorrhage. Stroke, defined as abrupt-onset neurologic dysfunction lasting more than 24 hours, is the third leading cause of death and the leading cause of serious, long-term disability in this country.In 2008, direct and indirect costs related to stroke are estimated to total $65.5 billion. Nearly 25% of patients who experience a stroke have sustained a previous stroke, making secondary prevention of recurrent stroke an important target of pharmacotherapy.
A new population-based study shows that transient ischemic attack (TIA) precedes an acute stroke in only 12.4% of cases, ranging up to about 20% for large artery strokes.
The results suggest more resources for primary prevention strategies should be considered over urgent-care TIA clinics that will prevent only "a small but significant fraction of the current stroke burden," The risk for stroke within 3 months of a TIA is estimated to be about 17%,
Trials of urgent evaluation and care for patients who have experienced TIA have shown that early and aggressive intervention can significantly cut this risk. For example, results of the Early Use of Existing Preventive Strategies for Stroke (EXPRESS) trial, of which Dr. Rothwell was also principal investigator, showed urgent intervention cut the 90-day risk for recurrent stroke by 80%, as well as reducing fatal and nonfatal stroke, disability, hospital admission days, and costs by the same magnitude
Ischemic strokes can be generally classified as cardiogenic or noncardiogenic in origin. Cardiogenic strokes are caused by release of blood clot emboli that originate from cardiovascular conditions such as atrial fibrillation, valvular heart disease, and severe left ventricular dysfunction. Emboli can travel to the brain and occlude cranial arteries, resulting in ischemia.
Noncardiogenic strokes and transient ischemic attacks (TIAs) result from atherosclerotic plaques within either intracranial or extracranial arteries. Similar to a myocardial infarction, these plaques can rupture, causing collagen exposure, platelet aggregation, and clot formation. The ruptured plaque can either occlude the artery locally or break off, resulting in emboli that block arteries deep within the brain.
The American Heart Association (AHA) and the American Stroke Association (ASA) published joint guidelines in 2006 for treatment of patients with a history of noncardioembolic stroke or TIA.The guidelines recommend antiplatelet agents for secondary prevention of ischemic stroke or TIA. Specifically, monotherapy with aspirin, the combination of extended-release dipyridamole plus aspirin, and monotherapy with clopidogrel are all considered acceptable first-line options for antiplatelet therapy.
Of these options, aspirin monotherapy has been the mainstay of treatment for secondary prevention of ischemic stroke or TIA. It has been studied extensively and is relatively safe and inexpensive. Unfortunately, aspirin monotherapy has its limitations. It has been suggested that aspirin alone produces only a 10-15% relative risk reduction in stroke recurrence compared with placebo.
Aspirin can also cause significant gastrointestinal discomfort and bleeding, and certain patients may be resistant to its antiplatelet effects. In clinical studies, patients treated with aspirin who had aspirin resistance, determined by a variety of aspirin resistance assays, were at increased risk for stroke, myocardial infarction, and other cardiovascular events compared with patients without aspirin resistance. These findings have led investigators to consider alternatives to aspirin monotherapy, most notably, combination antiplatelet treatment.
Clopidogrel acts on platelets by irreversibly binding the adenosine diphosphate (ADP) receptor, blocking the ADP-dependent activation of the glycoprotein IIb-IIIa complex. This complex works as a receptor for fibrinogen on the surface of the platelet.
Dipyridamole inhibits the uptake of adenosine into platelets, resulting in elevated local adenosine concentrations. Adenosine then acts on platelet A2 receptors, increasing the production of cyclic adenosine monophosphate. This mechanism prevents platelet-activating factor, collagen, ADP, and other stimuli from activating platelet aggregation.By blocking platelet aggregation through multiple mechanisms, it is postulated that secondary ischemic stroke prevention can be enhanced.
over 6600 patients with TIA or stroke in the past 3 months to one of four regimens: aspirin 25 mg twice/day plus extended-release dipyridamole 200 mg twice/day, aspirin 50 mg/day alone, extended-release dipyridamole 200 mg twice/day alone, or matching placebo.Roughly 35% of patients had ischemic heart disease, 22% had peripheral vascular disease, and 14.5% had diabetes mellitus.
Combination Antiplatelet Agents for Secondary Prevention of Ischemic Stroke
Primary end points were stroke, death, and composite of stroke or death. Secondary end points included the frequency of TIA. After a mandatory follow-up period of 2 years for all patients, the combination of aspirin 25 mg twice/day plus extended-release dipyridamole 200 mg twice/day was found to have significantly reduced the risk of the primary end point of stroke by 37% (p<0.001) compared with a reduction of 18% with aspirin alone (p=0.013) and 16% with dipyridamole alone (p=0.039) versus placebo.Bleeding episodes were reported by 8.2% of those given aspirin alone, 8.7% of the group given dipyridamole plus aspirin, 4.7% of the dipyridamole monotherapy group, and 4.5% of the placebo group. Of these bleeding episodes, similar percentages were described as moderate to severe in the aspirin and the aspirin-dipyridamole groups.
After a mean follow-up of 1.9 years, 939 (9.8%) of patients in the clopidogrel group experienced primary composite events compared with 1021 (10.7%) in the aspirin group. This resulted in an 8.7% relative risk reduction (95% confidence interval [CI] 0.3-16.5, p=0.043) for the primary composite end point, favoring clopidogrel. In terms of bleeding, intracranial hemorrhage or any bleeding disorder occurred similarly with clopidogrel versus aspirin, whereas gastrointestinal hemorrhage occurred more frequently in patients taking aspirin than in those taking clopidogrel.
Thus, data from CAPRIE suggest that clopidogrel may be more effective than aspirin for secondary prevention of ischemic stroke, especially in patients who also have other forms of atherosclerotic vascular disease.
Since clopidogrel exerts its antiplatelet activity by selectively and irreversibly inhibiting binding of ADP to its platelet receptor, and aspirin exerts its antiplatelet activity by preventing formation of thromboxane A2, the combination of aspirin and clopidogrel was hypothesized to be superior to clopidogrel monotherapy in preventing secondary events.
To test this hypothesis, MATCH, a randomized, double-blind, placebo-controlled trial of 7599 patients, was undertaken. The researchers evaluated patients with a history of ischemic stroke or TIA within the past 3 months, plus one additional cardiovascular risk factor (i.e., previous stroke, previous myocardial infarction, angina, diabetes mellitus, or symptomatic peripheral artery disease).
The decision to study a relatively high-risk secondary prevention population was based on several posthoc analyses of the CAPRIE trial suggesting that the benefit of clopidogrel over aspirin was most evident in patients with a history of ischemic events, diabetes, or cardiac surgery.[25-27] For the same reason, clopidogrel 75 mg/day served as the comparator group versus clopidogrel 75 mg/day plus aspirin 75 mg/day.[19]
At baseline, 80% of patients were receiving aspirin, and 79% had an ischemic stroke as the qualifying event. Nearly 27% had an ischemic stroke and 19% had a TIA before the qualifying event. Other pertinent atherosclerotic vascular disease findings included 5% with a previous myocardial infarction, 10% with symptomatic peripheral artery disease, 13% with angina, and 68% with diabetes mellitus.
Patients were evaluated for the composite end point of ischemic stroke, myocardial infarction, vascular death (including hemorrhagic death of any origin), or rehospitalization for acute ischemia. Secondary end points were the individual outcome of stroke and bleeding, including minor, major, or life-threatening bleeding.
After a mean follow-up period of 18 months, the combination of aspirin plus clopidogrel failed to reduce the primary outcome of recurrent ischemic events over clopidogrel alone (relative risk reduction 6.4%, 95% CI -4.6-16.3, p=0.244). The individual secondary end points of ischemic stroke or any stroke were also not significantly different between treatment groups. Differences between therapies in several prespecified patient subgroups were also evaluated.
The presence or absence of diabetes, peripheral artery disease, previous ischemic stroke, TIA, or myocardial infarction did not create a statistically significant difference between treatments. Further, the risks of minor, major, and life-threatening bleeding were each significantly increased in the aspirin clopidogrel group compared with aspirin monotherapy. Overall, there were nearly 3 times more reported bleeding events in the combination group than in the aspirin monotherapy group.
Data from these historic antiplatelet clinical trials serve as the foundation for the current AHA-ASA and ACCP guidelines. Results from recent combination antiplatelet trials have increased our knowledge of antiplatelet therapy for secondary prevention of ischemic stroke.
Conclusion
Given current clinical trial evidence, extendedrelease dipyridamole plus aspirin has superior efficacy over aspirin monotherapy for secondary prevention of ischemic stroke or TIA. Outside of efficacy, patient comorbidities and economic factors should continue to guide selection of antiplatelet therapy. The combination of aspirin and clopidogrel should be reserved for those who have had coronary artery stenting. Clopidogrel monotherapy is the preferred treatment in patients with aspirin allergy or intolerance, and aspirin monotherapy is preferred when financial concerns arise.
• Four features that reliably predicted the diagnosis of ICH in patients with mild stroke, which together comprised the SCAN tool:
• severe hypertension (blood pressure 180/110 mm Hg at onset),
• confusion at onset,
• previous use of anticoagulants, and
• nausea or vomiting at onset.
• In the derivation cohort, at least one of these predictors was present in all patients with ICH, and 42% of patients with 2 predictors had ICH on imaging. On the other hand, the absence of any of the four variables essentially ruled out ICH (present in only 0.2% of patients without any of the variables).
The FDA has approved onabotulinumtoxin A for treating upper limb spasticity The US Food and Drug Administration (FDA) has approved a new indication for onabotulinumtoxin A injection (Botox; Allergan, Inc) for upper limb spasticity. This condition commonly occurs in the flexor muscles of the elbow, wrist, and fingers for adults with conditions such as stroke, traumatic brain injury, cerebral palsy, or progressive multiple sclerosis.
The drug is derived from Clostridium botulinum and serves to inhibit acetylcholine release, leading to a temporary paralysis of the spastic muscle.
The condition can occur after stroke or spinal cord/traumatic brain injury and also result from cerebral palsy or progressive multiple sclerosis.
Treatments with onabotulinumtoxin A should be spaced at least 12 weeks apart. For spasms in the biceps brachii, 100 to 200 units should be distributed over 4 sites; for the flexor carpi radialis/ulnaris muscles, 12.5 to 50 units in 1 site should be administered; and for the flexor digitorum profundis/sublimes, 30 to 50 units in 1 site should be given.
In clinical trials, adverse events most commonly included nausea, fatigue, bronchitis, muscle weakness, and arm pain. Potentially fatal swallowing and breathing symptoms may occur hours to weeks after injection. Other botulism symptoms may include asthenia, generalized muscle weakness, diplopia, blurred vision, and ptosis.
Contraindications to treatment include hypersensitivity to any botulinum toxin preparation or formulation components, and infection at the proposed injection site.
The FDA last year implemented a boxed warning for the product regarding the risk of spreading of botulinum toxin from the injection site to other parts of the body, causing botulism symptoms such as asthenia, generalized muscle weakness, diplopia, blurred vision, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence, and breathing difficulties.
These symptoms have been reported hours to weeks after injection; swallowing and breathing difficulties can be life-threatening, and fatalities have been reported.
Because of the risk for potentiation of toxin effects, caution is advised with concomitant use of aminoglycoside antibiotics or other agents that interfere with neuromuscular transmission such as curare-like compounds; anticholinergic effects may be increased by use of additional anticholinergic agents.
Onabotulinumtoxin A previously was approved for the treatment of severe primary axillary hyperhidrosis, blepharospasm, strabismus, cervical dystonia, and cosmetic purposes.
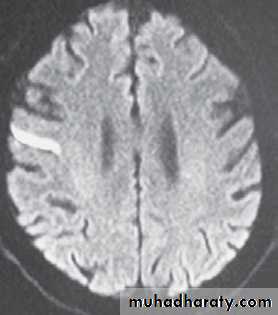


A 72-year-old man presented with a sudden onset of slurred speech. His medical history was unremarkable, and he was taking no medications and did not smoke. The physical examination revealed left-sided facial paralysis and dysarthrophonia. Diffusion-weighted magnetic resonance imaging showed a hyperintensity in the right precentral gyrus, reflecting acute cerebral ischemia (Panel A, arrow). Three days after the stroke, the patient showed impaired voluntary innervation. When he was asked to smile on command, he was unable to
fully smile because of facial paralysis on the left side (Panel B). However, during emotional encounters, the patient was able to overcome the facial paralysis (Panel C).It is hypothesized that this dissociation of emotional and volitional facial movement is due to separate origins of corticofacial projections. The nerve tracts affecting
voluntary facial movement probably originate from the main motor cortex. Those affecting involuntary movement during emotion probably arise from the audal cingulate motor cortex, a medial brain region with inputs from the limbic system
Copyright © 2010 Massachusetts Medical Society.
Pseudoathetosis
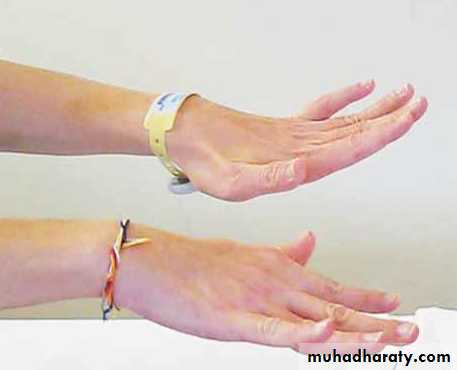
This pattern of movement
is known as “pseudoathetosis.” It is clinically indistinguishable from true athetosis, but pseudoathetosis is caused by loss of proprioception, whereas athetosis is due to structural abnormalities in certain areas of the brain. Pseudoathetosis has been reported in spinal cord disorders but more often occurs in disorders of the central or peripheral nervous system. In this patient, magnetic resonance imaging of the brain and spinal cord and extensive studies of serum and cerebrospinal fluid produced unremarkable results. He was given a diagnosis of idiopathic sensory ataxic neuropathy and received a course of intravenous immune globulin, with no effect. Methylprednisolone was then administered for 5 days, followed by a short tapering course of oral glucocorticoids. He showed a partial recovery 5 months after the initial presentation. At 11 months, there was no further abatement of neurologic signs.Copyright © 2010 Massachusetts Medical Society
A n otherwise healthy 40-year-old man presented with a 3-week history of paresthesia and inability to control his hands. Physical examination revealed generalized areflexia, marked loss of position sense, loss of feeling in a glove-and-stocking distribution, and preserved motor strength. The patient also had
a broad-based gait and dysmetria in all four limbs. Romberg’s sign was present. When he stretched out his arms, his fingers showed involuntary, constant, slow writhing movements that became more prominent with eye closure. The movements could also be seen in his toes, although to a lesser extent
• Results from several pivotal studies have contributed to our knowledge of stroke. Additional data support the efficacy and safety of intravenous alteplase, the standard of care for acute ischemic stroke since 1995. Due to these study results, the American Stroke Association changed its recommendation to extend the time window for administration of intravenous alteplase from within 3 hours to 4.5 hours of symptom onset; this recommendation enables many more patients to receive the drug.
Other findings included clinically useful biomarkers, the role of inflammation and infection, an expanded role for placement of intracranial stents, a reduced role for urgent carotid endarterectomy, alternative treatments for large-vessel disease, identification of nontraditional risk factors, including risk factors for women, and newly published pediatric stroke guidelines.
In addition, new devices for thrombolectomy are being developed, and neuroprotective therapies such as the use of magnesium, statins, and induced hypothermia are being explored. As treatment interventions become more clearly defined in special subgroups of patients, outcomes in patients with acute ischemic stroke will likely continue to improve.
• Stroke is the number one cause of adult disability in the United States and Europe and is the third leading cause of death in the United States. Approximately 15% of strokes are hemorrhagic and 85% are ischemic.
• Acute ischemic stroke (AIS) is a heterogeneous group of vascular diseases that encompasses large-artery atherosclerosis (16.3%), penetrating small-artery disease (lacunar infarcts, 15.9%), cardiogenic embolism (29.1%), stroke of unknown etiology (36.1%), and stroke of other determined etiology (2.6%).
A stroke may occur in the arterial or venous vasculature and may be due to either intracranial or extracranial disease.
Large-artery strokes may result from atherogenic embolus or hypoperfusion. These strokes may manifest with large clot burdens and more severe baseline neurologic deficit and, as a consequence, may fail traditional AIS interventions. Malignant middle cerebral artery (MCA) occlusions represent a special subtype of large-artery strokes, the manifestation of which differs from that of other AIS cases.
Recent clinical trials focusing on alternative interventions have provided new information about successful recanalization strategies in large-vessel occlusions.
Cardiogenic embolism may result from an atrial or ventricular thrombus as a consequence of atrial fibrillation, atrial flutter, mechanical or prosthetic heart valves, recent myocardial infarction, or cardiomyopathy. Iatrogenic causes of cardiogenic stroke include cardiac catheterization, coronary artery bypass surgery, percutaneous transluminal coronary angioplasty or valvulplasty, intraaortic balloon pump, or cardiac transplantation.
Lacunar infarcts are distinctive in the stroke classification scheme because they do not require demonstration of a vascular lesion. Also, these infarcts may be the result of hypertension, diabetes mellitus, dyslipidemia, or genetic disorders, such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencepahlopathy (CADASIL), which is an autosomal dominant mutation on the NOTCH3 gene in chromosome 19 that results in thickening of the muscular wall of small blood vessels typically in white matter of brain.
Stroke of other determined etiology includes nonatherosclerotic vasculopathies, prothrombotic states, dissections, paradoxic embolization through a patent cardiac septal defect, illicit drug use, and autoimmune disorders. This classification of AIS cases includes those with multiple causes or no probable evidence to be able to establish a single cause.
Viral infections, most notably influenza, in addition to bacterial infections appear to be an inflammatory trigger that may precede up to one third of ischemic strokes. The highest stroke risk appears to occur within 1 week of an acute infection, and the severity and clinical outcome of AIS may be worse when preceded by an infection. Both acute and chronic infections may increase inflammation biomarkers such as C-reactive protein and interleukin-6 and may represent future areas of targeted therapies for AIS in the presence of infection. Other inflammatory biomarkers including interleukin-6, tumor necrosis factor–α, C-reactive protein, and fibrinogen are associated with recurrence of stroke.
• Rapid recognition of stroke symptoms is paramount because "time lost is brain lost," and several interventions require strict adherence to timelines that begin at symptom onset. The five cardinal signs of stroke include
• weakness,
• speech impairment,
• vision impairment,
• headache, and
• dizziness.
• Vision impairments commonly involve a sudden loss of vision usually in one eye or double vision. Sudden severe and unusual headache (often referred to as a "thunderclap" headache), described as the worst headache of one's life, is most often associated with hemorrhagic stroke (particularly subarachnoid hemorrhage). Dizziness may be accompanied by sudden loss of balance and may result in a fall.
• Stroke Centers
• The AIS stroke guidelines adopted the recommendation by the Brain Attack Coalition for the establishment of two designations of stroke centers: primary stroke centers and comprehensive stroke centers. The Florida Stroke Act of 2004 requires that patients experiencing a stroke be transported by emergency medical services to a certified stroke center.One recent study applied a pragmatic stroke system model to improve outcomes of intravenous alteplase therapy in a community hospital supported by a regional stroke system.[24] The authors reported that these outcomes at the stroke center showed a reduced rate of symptomatic intracranial hemorrhage of 3.3% compared with 6.4% in emergency department settings in the original National Institute of Neurological Disorders and Stroke (NINDS) Recombinant Tissue Plasminogen Activator (rt-PA) Stroke Trial.
In addition, it was reported that 3-month modified Rankin Scale (mRS; a scoring system for measuring disability) scores of 0–1 (indicating no to minimal disability; scores of 5–6 indicate severe disability or death) were 54% versus 43% in the NINDS rt-PA trial (p=0.04). Mortality rates were 13% at the stroke center versus 17% in the NINDS rt-PA trial.
In addition to stroke centers, teleneurology systems are being developed to reach patients with AIS in rural areas who may not otherwise have access to a stroke neurologist
• Patient Evaluation and Management in the Emergency Department
• According to data published by the Paul Coverdell National Acute Stroke Registry, nearly 50% of patients in the registry who experienced a stroke came to an emergency department within 2 hours of symptom onset. Thus, the establishment of protocols for the emergency triage and management of patients with acute stroke is critical.[3] Patients should be evaluated and decisions on treatment made within 60 minutes of patient arrival to the emergency department.Patients must receive airway, breathing, and circulatory assessment followed by a neurologic evaluation.
Determining the time of onset of stroke symptoms is imperative. Patient assessment with use of the National Institutes of Health Stroke Scale (NIHSS)[25, 27] and mRS[25] may be conducted by nurses certified in this process to determine stroke deficits .The NIHSS evaluates neurologic impairment on a scale of 1–42, with higher scores indicating severe neurologic impairment and lower scores less severe impairment.
Any baseline score other than zero warrants neurologic work-up. These standardized scales may provide outcome assessments when performed at presentation, 24 hours after admission, and again at discharge. All patients with stroke must be screened for dysphagia. Failure to screen may result in oral drugs being inappropriately administered and result in aspiration pneumonia.
• As part of the acute stroke pathway, a number of diagnostic tests should be performed including blood glucose level, serum electrolyte level, complete blood cell count, platelet count, renal function studies, prothrombin time, activated partial thromboplastin time, oxygen saturation, and cardiac markers.[3] Patients should receive continuous oxygen therapy with oxygen saturation and cardiac monitoring.
Those presenting with a cardiac history should undergo electrocardiography since patients with stroke of thromboembolic origin may have acute myocardial infarction as well; however, management of these patients is outside the scope of this article.
• Biomarkers
• The role of plasma biomarkers in AIS is under investigation. N-Methyl-D-aspartate receptor autoantibodies may be detectable in plasma during AIS and play a vital role in the ischemic cascade.[28] Other proteins involved in brain ischemia include astroglial protein S100B, B-type neurotrophic growth factor, von Willebrand factor, and matrix metalloproteinase (MMP)-9. Inflammatory biomarkers associated with the acute-phase response of AIS include interleukin-6, tissue necrosis factor-α, C-reactive protein, and fibrinogen.[19] One predictive biomarker model included brain natriuretic peptide, C-reactive protein, D-dimer, MMP-9, and S100B, and a sensitivity of 81% and specificity of 70% for diagnosing AIS were reported.[7]Biomarkers such as MMP-9 and ferritin may have additional utility in predicting hemorrhagic complications after fibrinolytics and in identifying patients at risk for developing complications.[7, 29] The presence of plasma MMP-9 is known to play a deleterious role in AIS due to its ability to degrade laminin and fibronectin, important components of the blood-brain barrier.
Animal studies show that during cerebral ischemia, free iron is released from intracellular stores and catalyzes the conversion of superoxide and hydrogen peroxide into a highly toxic hydroxyl radical contributing to higher oxidative stress and inflammation.[29] In humans, baseline ferritin levels greater than 79 ng/ml before administration of intravenous alteplase increased hemorrhagic transformation and cerebral edema.[29] The presence of plasma MMP-9 and high ferritin levels have been demonstrated to be an independent predictor of hemorrhagic transformation in patient with AIS.
Urgent brain imaging should be conducted with use of computed tomography (CT) without contrast material enhancement, which is the current gold standard used to differentiate hemorrhagic causes of neurologic injury from nonvascular injury to the brain. This provides the clinician with information regarding size, location, and vascular distribution of the infarct.
The differential diagnosis for AIS includes ruling out migraine, hemorrhagic stroke, head trauma, brain abscess, encephalitis, brain tumor, seizure with postictal paralysis, and hypoglycemia.
• Goals of Therapy
• For all interventions performed urgently in patients with AIS, the goals are limiting the area of ischemia (infarct) and salvaging the penumbra. The penumbra is a hypoperfused area of focal ischemia that is potentially viable and may be salvaged by timely and appropriate intervention, including maintenance of euvolemia. Once the patient is stable, the goal of AIS therapy is to remove the occlusion through recanalization. When contraindications to removing the occlusion are present, preventing extension of the infarct becomes critical.• Intravenous Alteplase
• The administration of rt-PA, a fibrinolytic agent used to remove the occlusion, remains one of the few class I, level of evidence A drug therapy recommendations in the treatment of AIS.[3] Administration should be considered only if the benefit outweighs the risk and no exclusion criteria exist.Absolute contraindications to the use of intravenous alteplase for acute ischemic stroke include the following:
Evidence of intracranial hemorrhage on pretreatment evaluation
Suspicion of subarachnoid hemorrhage on pretreatment evaluation
Recent (within 3 mo) intracranial or intraspinal surgery, serious head trauma, or previous stroke
History of intracranial hemorrhage
Uncontrolled hypertension at time of treatment (> 185 mm Hg systolic or > 110 mm Hg diastolic blood pressure)
Seizure at the onset of stroke
Active internal bleeding
Intracranial neoplasm, arteriovenous malformation, or aneurysm
Known bleeding diathesis including but not limited to the following:
Current use of oral anticoagulants (e.g., warfarin sodium) or an international normalized ratio greater than 1.7 or a prothrombin time greater than 15 seconds
Administration of heparin within 48 hours before the onset of stroke and an elevated activated partial thromboplastin time at presentation
Platelet count less than 100 x 103/mm3
• Alteplase, a tissue plasminogen activator, is an enzyme that can convert plasminogen to plasmin as a result of fibrin enhancement. At pharmacologic concentrations, it binds to fibrin within the thrombus, forming an active lytic complex. The recommended alteplase dose for patients with AIS is 0.9 mg/kg (maximum 90 mg), with a bolus of 10% of the dose administered over 1 minute, and the remainder infused over 60 minutes.
Significant drug interactions may occur with concomitant administration of anticoagulant and antiplatelet agents, both of which increase the bleeding risk. The greatest risk of treatment with alteplase is symptomatic intracranial bleeding.[3, 30] Therapeutic heparin, antithrombotics, and anticoagulants are contraindicated within 24 hours after administration of alteplase.
Mechanical prevention of deep vein thrombosis traditionally has been preferred over medical prevention; however, the recently conducted European Cooperative Acute Stroke Study (ECASS) III used subcutaneously administered heparin at a daily dose of 10,000 units or less without increased bleeding complications.[6]
Postmarketing studies report the presence of laryngeal and orolingual angioedema to be less than 1%, but this life-threatening situation may require urgent airway stabilization. An observational study found that the frequency of orolingual angioedema was 1.7% (95% confidence interval [CI] 0.2–5.9%) with angiotensin-converting enzyme (ACE) inhibitor therapy as a risk factor.[31] As plasminogen is converted to plasmin, the plasmin cleaves bradykinin from high-molecular-weight kininogen.[32] This coupled with an ACE-inhibitor–mediated decrease in bradykinin metabolism along with increased neurokinin levels might explain the increased risk.
• The NINDS rt-PA trial established safety and efficacy of alteplase administered within 3 hours of symptom onset; this study has become the gold standard to which all other intravenous fibrinolytic AIS studies are compared.[33] However, a delay in reporting of symptoms and transport to an emergency department are the largest hindrances to the administration of intravenous alteplase.
The recently published results of the ECASS III study showed the safety and efficacy of an expanded physiologic time window from 3 to 4.5 hours in patients with mild-to-moderate AIS.[6] This study, in light of other clinical trials, a recent meta-analysis, and stroke registry data, resulted in the AHA-ASA recommendation to expand the time window from 3 to 4.5 hours in a valid AIS population.[34] In addition to the expanded time window, the AHA-ASA cautions that application of similar exclusion criteria must be used in the clinical setting, citing no proven benefit in patients excluded from the ECASS III study.[34]
The committee made another recommendation cautioning that the expanded time window should not be interpreted as a reason to relax the sense of urgency with patients with AIS.[34]
• Efficacy
Final analysis indicated that treated patients were at least 30% more likely than those in the placebo group to have minimal or no disability at 90 days.• Safety
The NINDS rt-PA study reported that the most severe adverse event was symptomatic intracranial hemorrhage occurring within 36 hours of symptom onset in 6.4% of patients treated with alteplase versus 0.6% in those receiving placebo (p<0.001).[33] Intracranial hemorrhage was considered symptomatic if it was not previously seen on CT scan and it resulted in any decline in neurologic status, strict adherence to exclusion and inclusion criteria, and appropriate blood pressure management may decrease occurrence of hemorrhage.
The A0276g trial assessed efficacy and safety of intravenous alteplase in patients treated between 0 and 6 hours after onset of stroke symptoms.[35] This trial demonstrated that intravenous alteplase administered 5–6 hours after symptom onset in patients with moderately severe–to-severe strokes may cause harm.
• Intravenous and Intraarterial Alteplase Combination Therapy
• Intraarterial fibrinolytics may be used with a mechanical clot removal device or may be used independently. The 2007 AIS guidelines added the recommendation that interventions to restore perfusion cannot be recommended outside the setting of clinical trials (class III, level B). A pilot study, the Interventional Management of Stroke Part 1 (IMS 1), used low-dose intravenous alteplase combined with intraarterial alteplase with better efficacy results and similar intracranial hemorrhage results compared with those results in the NINDS rt-PA study.[46]The Interventional Management of Stroke Part 2 (IMS 2), a second pilot study, used a similar protocol of intravenous alteplase 0.6 mg (bolus of 15% dosed over 1 min and remainder infused over 30 min) within 3 hours of symptom onset.[47] Patients were transferred for immediate angiography, and those with a visualized occlusion received intraarterial alteplase in doses up to 22 mg within 5 hours of symptom onset with completion within 7 hours of symptom onset. The baseline median NIHSS score was 19 (inclusion criterion was a minimum NIHSS score of 10) and the median time of treatment was 142 minutes compared with 108 minutes for placebo (p<0.001).
One difference between IMS 1 and IMS 2 was the utilization of the EKOS micro-infusion system (EKOS Corp., Bothell, WA), a low-energy ultrasound device that may reversibly alter the structure of the thrombus and accelerate thrombolysis. Before initiation of intraarterial therapy, an intravenous heparin 2000-unit bolus followed by an intravenous heparin infusion maintained at 450 units/hour was administered and was discontinued at completion of the intraarterial therapy.
• Mechanical Thrombectomy and Fibrinolytics
• In 2007, the AIS stroke guidelines recognized the Merci Retriever (Concentric Medical, Mountain View, CA) as a mechanical thrombectomy device with benefits in carefully selected patients with AIS.[3] Because recanalization is less likely after intravenous alteplase in patients with large-vessel AIS as a consequence of large-clot burdens, thrombectomy offers an alternative method of recanalization in this population.[3, 52] In addition, this approach may be more desirable in patients ineligible to undergo fibrinolysis because of recent surgery. However, this intervention must be performed in a catheterization laboratory by experienced radiologists skilled in neuroendovascular techniques.
• Urgent Carotid Endarterectomy
• generally is not performed in patients with AIS because the sudden restoration of blood flow may increase the development of brain edema or lead to hemorrhagic transformation, especially among patients with major infarctions. A recently published systematic review of studies published between 1980 and 2008 reported the pooled absolute risk of stroke or death after urgent carotid endarterectomy was 20.2% (95% CI 12.0–28.4%) and was greater than that in patients with stable disease who undergo carotid endarterectomy (OR 1.2, 95% CI 0.9–1.6, p=0.13).• Intracranial Stent Placement
• Until recently, intracranial stent placement was limited to off-label use of balloon-mounted stents designed for cardiac circulation.[11] Because these stents are rigid, they are poor tools for treating intracranial disease because of weak navigational ability in the tortuous intracranial circulation.[11] A new generation of self-expanding intracranial stents as an option for patients refractory to conventional AIS management offers new alternatives for intracranial stent placement.The Wingspan self-expanding intracranial stent (Boston Scientific, Natick, MA) has demonstrated technical feasibility with high rates of recanalization. [11] Optimal long-term safety strategies to limit in-stent thrombosis must be determined before large clinical trials can be designed since subacute stent thrombosis has occurred in patients with standardized antiplatelet therapy.[56]
• The Heparin Controversy
• The heparin controversy revolves around studies in which heparin was used inappropriately (albeit retrospectively) and resulted in poor outcomes. A consensus among stroke management providers is that the efficacy of intravenous unfractionated heparin (UFH) has been inadequately tested in patients with defined stroke subtypes and occlusive vascular lesions.[57] In the presence of contraindications to clot removal, UFH may be useful in the treatment of AIS in patients with specific stoke subtypes. These subtypes include cerebral venous thrombosis, large-artery occlusions with a critical stenosis, or cardiac sources of emboli. Unfractionated heparin may be used as a bridge to full therapeutic anticoagulation with warfarin when warfarin is indicated.[58, 59]The current AIS guidelines recommend against the use of heparin in patients with AIS regardless of the underlying issue (class III, level A), whereas the American College of Chest Physicians guidelines recommend UFH for patients with AIS of cardioembolic sources.[3, 60] When UFH is used in patients with AIS, blood pressure must be managed appropriately to reduce the risk of hemorrhagic transformation. Monitoring for heparin toxicity should include platelet counts for early identification of heparin-induced thrombocytopenia.
• One thing remains consistent between the guidelines and stroke management providers: there is no role for therapeutic dosing of low-molecular-weight heparin (LMWH) in the treatment of AIS.[3] A meta-analysis evaluated safety and efficacy data from 10 randomized controlled trials of LMWHs in patients with AIS.[61] Although LMWHs reduced venous thromboembolic events in patients with AIS, the risk of extracranial bleeding did not outweigh the reduction in death or disability. Therefore, the consensus among stroke management providers is that LMWH has no role in the treatment of AIS, but it may be used along with heparin to prevent deep vein thrombosis.[3]
• Antithrombotic Agents
• Aspirin 325 mg/day should begin within 24–48 hours of AIS therapy and is the only anti-thrombotic to receive a class I, level A rating.[3] Because previous stroke is a risk factor for another AIS event, patients may be taking aspirin, clopidogrel, or extended-release dipyridamole–aspirin at presentation. Although these agents should not be given within 24 hours of intravenous alteplase, their previous use does not preclude administration of intravenous alteplase. The standard of care is to determine compliance with the presenting agent, and if the patient is compliant, a change to another agent may be considered or alternatively the addition of low-dose aspirin to clopidogrel may be considered for breakthrough therapy.Maintenance of Euglycemia
Hyperglycemia has been associated with poor outcomes, and an increase in blood glucose level is an indicator of a more serious stroke. It may affect the blood-brain barrier, causing brain edema and possible hemorrhagic transformation.[62] In 728 patients both diabetic and nondiabetic from the ECASS II trial, glucose levels were measured at baseline and at 24 hours.[63] Results showed that nondiabetic baseline hyperglycemia was not associated with worsening outcomes, but persistent hyperglycemia was associated with adverse events.
In patients with diabetes, hyperglycemia was not related to stroke outcomes. The recently published Normoglycemia in Intensive Care Evaluation–Survival Using Glucose Algorithm Regulation (NICE-SUGAR) trial evaluated intensive lowering of blood glucose level (range 81–108 mg/dl) compared with conventional lowering (< 180 mg/dl) in critically ill patients.[64]
The intensive group showed deleterious outcomes in critically ill patients, whereas a lower mortality was linked to the conventional group. A meta-analysis of five randomized controlled trials revealed that although intensive control of hyperglycemia decreases cardiovascular events, it does not have an effect on all-cause mortality.[65] Controversy still exists as to which patients should receive intensive control as well as the appropriate goal level for blood glucose. Concerns of the patient developing hypoglycemia are valid and should be addressed with frequent monitoring of blood glucose levels.
Blood Pressure Management
Assessment and management of blood pressure during AIS are critical. Many patients experiencing stroke will have a natural decline in blood pressure during the first 24 hours after the event and may become hemodynamically unstable, a negative prognostic risk factor.[66, 67] The causes of hypotension must be evaluated and treated. Treatment for hypovolemia includes volume replacement with normal saline.[3] Vasopressor support with dopamine may be used.[3]Often, patients with acute strokes will have an increase in blood pressure, which may represent a compensatory mechanism to maintain cerebral blood flow. Other causes of hypertension include pain, nausea, acute stress due to the event, increased bladder pressure, and increased intracranial pressure.[67] Pain control with nonsedating drugs such as topical anesthetic patches or ice packs to localized areas of pain may be preferred in order to avoid sedation and interference with neurologic assessment.
Blood pressure goal setting must be individualized to the patient. Patients receiving intravenous alteplase require a systolic blood pressure less than 185 mm Hg and diastolic blood pressure less than 110 mm Hg to avoid hemorrhagic transformation.[3] For patients not receiving fibrinolysis, a reduction in blood pressure should be avoided to prevent a decrease in cerebral blood flow and cerebral perfusion pressure, which may expand the infarct and cause harm.
Because long-standing hypertension upregulates the cerebral perfusion pressure oxygenation curve, high blood pressure at presentation should not be treated unless a compelling reason exists such as concurrent myocardial infarct, hemorrhagic transformation, or other end-organ damage.[67] Furthermore, abrupt reductions in blood pressure must be avoided. Blood pressure agents of choice are those that are easily titratable and have safe and predictable results.[3] First-line therapies include nicardipine and labetalol.[3] Nicardipine's advantage is easy dose titration and predictability. It can be rapidly titrated, and after discontinuation the blood pressure returns to baseline.
Labetalol's advantages include its low cost and β-blockade in patients with atrial fibrillation; it represents a rational first-line agent for patients who cannot take oral agents and those who have been taking β-blockers before the event, thereby avoiding abrupt withdrawal of the drug. Each patient's comorbidities and current drug therapies should be assessed before deciding on an appropriate agent. Nitroprusside is now considered a third-line agent because of its undesirable safety profile, as it may contribute to increased intracranial pressure and cyanide toxicity.
• Neuroprotectants
• Many neuroprotective drugs for AIS have appeared to work in animals but fail when tested in humans.[69] The mechanism of action of neuroprotectants typically targets direct or indirect antagonism of glutamate, a known neurotoxic central nervous system transmitter. Indirect mechanisms include antagonism of N-methyl-D-aspartic acid (NMDA), a glutamate mimetic at the NMDA receptor, or inhibition of nitric oxide, a known stimulant of glutamatemediated processes. Once glutamate-mediated processes are started, highly toxic peroxynitrate free radicals are formed, causing brain cell apoptosis.Many neuroprotective therapies are being studied in patients with AIS. In addition to hypothermia, intravenous magnesium has shown promise. Statins and cerebral edema containment are also being evaluated.
Statins
The benefit of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) in patients with AIS may be independent of lowering the levels of low-density lipoprotein cholesterol. Pleiotropic effects include plaque stabilization and antiinflammatory effects; a review of the pleiotropic effects of statins is outside the scope of this article.The Northern Manhattan Observational Stroke Study (NOMAS) was a population-based study that identified a decline in clinical worsening in patients admitted for AIS to a large university medical center who were taking statins at presentation compared with those not taking a statin (5.3% vs 12.2%, p=0.04).[71] In addition, 90-day mortality was reduced in the statin group (1.8% vs 10.6%, p=0.03). The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial established the role of high-dose stains in secondary prevention; the Neuroprotection with Statin Therapy for Acute Recovery Trial (NeuSTART II) is a safety study evaluating shortterm, high-dose lovastatin use in patients with AIS.[72, 73] The pleiotropic effects of statins as a class effect and optimal dosing strategies remain unanswered and are being studied in clinical trials. Statin withdrawal in patients with AIS should be avoided.[74]
Magnesium
Magnesium blocks calcium-mediated glutamine reuptake into neuronal axons. The Field Administration of Stroke Therapy–Magnesium (FAST-MAG) open-label, single-arm pilot study allowed paramedics to administer intravenous magnesium 2.5 g in the field to patients with probable stroke followed by an additional 1.5-g bolus and maintenance infusion of 16 g delivered over 24 hours.[70] Patients were followed for 90 days.Although the trial had a small sample (20 patients), results indicated improvement in 20%, deterioration in 7%, and no change in 73% of patients receiving intravenous magnesium. At 3 months, 69% of patients had good functional outcomes, whereas 20% died. A large phase III clinical trial, the goal of which is to evaluate the effectiveness and safety of field-initiated magnesium sulfate in improving the long-term functional outcome of patients with acute stroke, is under way.[75]
Hypothermia
Induced hypothermia is one of the most promising neuroprotective therapies in AIS because increased body temperature has been associated with poor stroke outcomes.[9, 76] Patients with AIS most likely to benefit from this therapy include those with moderate-to-severe stroke and those with large-vessel occlusions and cerebral edema.[77] Also, elevated body temperatures immediately after stroke predict poor outcomes; therefore, controlling fever with acetaminophen and temperature-controlling devices should be a priority.A decreased body temperature to hypothermic levels of 32–36°C has proved neuroprotective in both humans and animal models.[77] Hypothermia slows metabolic demands on the body, decreases calcium influx into cells, and suppresses the production of free radicals. Such methods have been proved useful in patients with cardiac arrest at presentation; therefore, the same principles may be applied to patients with stroke.[78] The 2007 guidelines recommend induced hypothermia in patients with AIS as a class III, level B recommendation.[3]
Cooling can be divided into three distinct phases: induction, maintenance, and rewarming. Based on the available literature, temperature control should ideally start within 4–12 hours after symptom onset, and cooling should progress as rapidly as possible.[9] Care should be taken not to decrease body temperature below the target when inducing and maintaining temperatures.
The appropriate duration of hypothermia has not been established, and the number of patients treated with hypothermia in controlled trials is small. However, one trial maintained patients at an average of 32°C for 48 hours (range 12–72 hrs) with safe and effective outcomes.[77] Another study investigated hypothermia at 12 hours and 24 hours and found no significant difference in outcome.[79]
Methods of achieving hypothermia vary, but essentially they are either extravascular (topical) or endovascular. Extravascular methods include cooling blankets, ice baths and ice lavage, and conductive cooling-rewarming machines that supply continuous temperature-controlled water flow externally. Endovascular methods encompass the administration of chilled intravenous solutions often through a catheter placed into the central venous system usually by femoral, subclavian, or internal jugular access.[80] Multiple devices exist for both extravascular and endovascular cooling.
Clinicians should also be aware of potential adverse effects of therapeutic hypothermia including hypotension, infection, and cardiac arrhythmias. One of the most important measures to implement when inducing a hypothermic state is to sedate the patient because this will cause vasodilation and further facilitate heat loss through surface cooling.[9, 81] Monitoring parameters during cooling include continuous blood pressure and cardiac monitoring.[80] Hypothermia-induced bradycardia and hypokalemia may manifest when inducing hypothermic states.[9] Skin lesions, although rare, may be due to excessive exposure to cooling-device surfaces.[9]
A secondary temperature gauge in addition to the primary temperature gauge on the cooling device, such as a urinary bladder probe (only effective when the patient has adequate urinary output), a pulmonary artery probe, or rectal probe, may be used to adequately assess internal temperatures.[9] Patients are at a greater risk for developing
bacterial infections because inflammatory response is suppressed by cooling longer than 24 hours.
One major adverse effect of hypothermia is shivering, the body's natural way of warming.[9] Shivering expends energy and warms the body, both of which are undesirable during induced hypothermia. A number of countermeasures have been used to lower the shivering threshold such as warming the hands, feet, and face in addition to drugs such as buspirone, meperidine, magnesium, and neuromuscular blockade.[82] Neuromuscular blockade requires both mechanical ventilation and pain management and sedation.
Caution should be used when rewarming techniques are started. Monitoring parameters include continuous blood pressure and cardiac monitoring.[9] Rapid rewarming may produce rebound hypotension; therefore, a patient should not be rewarmed faster than a rate of 0.05°C/hour, and the warming process should take approximately 8–12 hours.[9] Rewarming must be slow and controlled to prevent adverse effects such as hypotension and hyperkalemia.[9, 83]
• Containment of Cerebral Edema
• Malignant MCA occlusions represent 10% of all AIS cases. Although most other patients with AIS will develop some cytotoxic edema, most will not develop high intracranial pressure as a consequence. Early-onset cytotoxic and vasogenic edema with sustained intracranial pressures greater than 20 mm Hg (normal is 5–15 mm Hg) may lead to permanent neurologic damage, brain herniation, or death. Mortality rates of up to 80% have been reported in this stroke subtype.When cytotoxic edema develops during a malignant MCA occlusion, it is within minutes to hours and does not respond to dexamethasone. Cytotoxic edema primarily reflects failure of the membrane sodium- and potassium-activated adenosine triphosphatase pump due to tissue ischemia and leads to early intracellular sodium, chloride, and calcium accumulation.[86]
The calcium and other stimuli trigger lipolysis, proteolysis, nitric oxide production, endonucleasemediated DNA degradation, and the activation of kinases and phosphatases.[86] These alter protein function and initiate upregulation of multiple genes. Such immediate early genes include those encoding transcription factors, heat shock proteins, cytokines, chemokines, and adhesion molecules.[86]
Vasogenic edema is characterized by progressive loss of blood-brain barrier integrity with extravasation of water, intravascular proteins, and inflammatory cells. Dexamethasone does have a role in vasogenic edema in brain hemorrhage, although its role is less clearly defined. Development of vasogenic edema tends to cause greater clinically significant space-occupying edema.[86]
Treatment of this subtype is aimed at containment of cerebral edema and includes decompressive hemicraniectomy, which is the surgical removal of part of the skull to accommodate the swollen brain.
The current 2007 AIS guidelines list hemicraniectomy as class IIa, level B,[3] but it has since gained popularity due to three recently published European randomized trials.[84, 85, 87] All three trials were stopped early due to improved outcomes in the hemicraniectomy group versus those receiving traditional medical management. Additional interventions are aimed at the management of intracranial pressure and are beyond the scope of this article.[88]
Ongoing Trials
Tirofiban, argatroban, desmoteplase, and tenecteplase are drugs currently being studied for the treatment of AIS.[89] Ongoing neuroprotective studies include those evaluating magnesium, minocycline, citicoline, and hypothermia.[89]Historically, AIS research has yielded little progress and lagged behind research in cardiac and other cardiovascular diseases.
Collaboration within the AIS community has increased over the years. Two such collaborative practices include the Stroke Therapy Academic Industry Roundtable (STAIR)[90] and the Specialized Programs of Translational Research in Acute Stroke (SPOTRIAS) consortium.[91]
The STAIR consortium brings together neurologists, other physicians, industry representatives, and regulators to discuss issues related to the development of new AIS therapies.[90] They recently recommended that clinical trials should focus on selected patient populations most likely to respond to investigational therapies. Additional recommendations include that penumbral imaging be incorporated into trials and that patients enrolled in investigational trials have rescue therapy options available when needed.
.
The National Institutes of Health (NIH) has been instrumental in the support of stroke studies and collaborative practices. The SPOTRIAS is an NIH-supported collaborative practice of eight centers that share resources and data to facilitate translation of basic research findings into clinical practice.[91] The development of specialized research resources, improved research model systems, and expansion of the research base through collaboration nationwide are methods to reaching the goal of reducing disability and mortality in patients with AIS.
Conclusion
As treatment interventions become more clearly defined in special subgroups of patients, outcomes in AIS will likely continue to improve. The heterogeneity of AIS has made targeted therapies more elusive. The past few years has yielded a plethora of new information relating to treatment of large-vessel disease and severe stroke, the role of biomarkers, infection and inflammation, thrombectomy, and intracranial stent placement.Intravenous alteplase has been the mainstay of AIS treatment since 1995, but the 3-hour time window between symptom onset and drug administration was often missed and patients were excluded. The landmark trial, ECASS III, identified patients with mild-tomoderate AIS who may safely receive intravenous alteplase up to 4.5 hours after symptom onset; this finding will enable many more patients to receive the drug. In addition, new devices for thrombolectomy are being developed, and neuroprotective therapies such as the use of magnesium, statins, and induced hypothermia are being explored
Synonyms: Tic Douloureuxa chronic, debilitating condition resulting in intense and extreme episodes of pain in the face. The episodes are sporadic and sudden and often like "electric shocks" lasting from a few seconds to several minutes.Trigeminal neuralgia results from a neuropathic disorder of the fifth cranial nerve (trigeminal nerve).
Trigeminal Neuralgia
The trigeminal nerve senses mixed modalities including:
Sensation
Nociception
Thermoception
Motor supply to the muscles of mastication
Most commonly the maxillary and/or mandibular branch are involved.
Epidemiology
Most commonly episodes occur after the age of 40.Annual incidence of about 4-5 per 100,0002 (however, these are based on strict case definitions and the true value may be almost six times higher).
More common in females.
There may also be a genetic predisposition as there have been observations of familial clustering. However, the exact method of transmission is unclear although there is a lack of penetrance.4
2-4% of patients will actually have multiple sclerosis.
Aetiology
Compression: blood vessels may press on the trigeminal nerve as it leaves the brain stem at its cerebellopontine nerve root. Compression of the nerve leads to demyelination. This results in spontaneous generation of electric impulses. This probably accounts for up to 90% of cases that were originally classified as idiopathic.Degeneration: some have postulated it to be part of the ageing process as with increasing age the brain atrophies leading to redundant arterial loops which can cause compression.
Myelin sheath infiltration e.g. tumour or amyloidosis.
Idiopathic
Presentation
There may be preceding symptoms e.g. tingling or numbness .Patients may have certain triggers that set the pain paroxysm off (see table below)
Followed by sharp, severe, shock like pains
These pains are usually on one side in the cheek or face but pain can involve the eyes, lips, nose and scalp
Episodes are intermittent but can last days, weeks or months on end and then not return for months or even years
3-5% of patients will have bilateral pains
Paroxysmal attacks of pain lasting a second to two minutes and affecting one or more divisions of trigeminal nerve (typically maxillary or mandibular branches)
Pain has at least one of the following characteristics: intense, sharp, superficial, stabbing, precipitated by trigger areas/factors
Attacks are similar in individual patients
No neurological deficit on examination
Not caused by another disorder
(Based on The International classification of headache disorders)
Diagnostic criteria for classic trigeminal neuralgia
Triggers of trigeminal neuralgia
VibrationSkin contact e.g. shaving, washing
Brushing teeth
Oral intake
Exposure to wind
Atypical trigeminal neuralgia
This subgroup of patients have relentless underlying pain like a migraine associated with superimposed stabbing pains. There may also be an intense burning sensation. This condition is particularly difficult to treat.
Differential diagnosis
Dental pathologyTemporomandibular joint dysfunction
Migraine
Temporal arteritis (TN rarely affects forehead alone)
Cluster headaches
Multiple sclerosis and other disorders of myelin
Overlying aneurysm of a blood vessel
Tumour in posterior fossa e.g. meningiomas
Arachnoid cyst at the cerebellopontine angle
Postherpetic neuralgia after shingles
InvestigationsThe diagnosis is clinical and it can be difficult to make. No investigations are required initially unless there is uncertainty regarding the diagnosis. Patients who are referred on for specialist review will usually have a brain MRI - this is to document the presence of compression and look for other intracranial causes of TN (5-10% of patients e.g. aneurysm, MS).5 There should be a lower threshold for earlier investigations in the following groups: younger patients, atypical symptoms, focal neurology and poor response to initial therapy.3
Management
Unfortunately there is no cure at present although newer surgical procedures are proving promising.Management involves three aspectsSupport and education
Medical
Surgical
Support and education
Patients need to be made aware that the condition is not life-threatening
Need however also to express empathy towards severity of the condition
Education as to the causes and potential therapies
Reassurance and support groups
Medical
Typical analgesics and opioid analgesics - these are unfortunately not very successful and they are thus not first line.Anticonvulsants e.g. carbamazepine or gabapentin. Carbamazepine and gabapentin are first line. Carbamazepine should be tried initially and the dose uptitrated to achieve pain control. If it fails to relieve the pain or adverse effects develop then try gabapentin.
However, there is a need for further randomised clinical trials to establish the effectiveness of these medications. These effects may be enhanced with baclofen and clonazepam - however, the efficacy is not well established and to date studies only involve a small number of patients.
Once patients have been in remission for 1 month the drug should be gradually withdrawn.
Tricyclic antidepressants e.g. low dose amitriptyline - the data supporting their use at present is lacking and CKS does not support their use.
Other drugs that might be used in a specialist setting include lamotrigine and baclofen.
Presence of atypical clinical features e.g. abnormal neurological signs.
Neither carbamazepine nor gabapentin are effective.Drugs cause unacceptable adverse effects even if pain relief is good.
Presence of atypical clinical features e.g. abnormal neurological signs.
Neither carbamazepine nor gabapentin are effective.Drugs cause unacceptable adverse effects even if pain relief is good.
Trigeminal neuralgia occurs in a person less than 40 years of age.
When and who to refer
Surgery
Most of the improvements in the management of trigeminal neuralgia have occurred because of advances in surgical treatments. Surgery involves either relieving pressure on the trigeminal nerve or damaging it to prevent any pain transmission.
Rhizotomy - the aim is to damage the trigeminal nerve. This is an alternative to the more invasive decompression. These methods include:
Percutaneous glycerol rhizotomy (under a local anaesthetic)
Percutaneous balloon compression rhizotomy (under a general anaesthetic)7
Radio-frequency rhizotomy (performed under sedation)
These procedures are good in the frail elderly patient or those with co-morbidities but unfortunately the affects are short lived and symptoms usually recur. They are usually associated with some level of sensory loss and can be repeated if necessary.
Stereotactic radiosurgery (gamma knife): - this is also a form of rhizotomy that uses radiation targeted at the trigeminal nerve root and thus injures it. Pain relief is usually delayed for a few days and there can be associated facial numbness. At present the number of locations providing this treatment is limited.3
NICE guidance on the use of stereotactic surgery in trigeminal neuralgia:8Surgery to be considered if severe pain or side effects from medication
A systematic review commissioned by NICE reported that between 33% - 90% achieved immediate pain relief with this procedure - only an average of 14% had recurrence of symptoms at 18 months
This requires a general anaesthetic. The approach is behind the ear into the posterior fossa on the affected side. Patients are usually assessed by MRI beforehand to look for the presence of compression.This procedure however is not with out risks. There is a risk of a cerebrovascular event, deafness and even death. The rates of these are dependant upon the surgeons expertise. However, it is associated with the best chance of long-term pain relief. The pain relief may not occur for a few weeks and there is little sensory loss.
Microvascular decompression:
Success rates with all surgical procedures are generally good with almost 3/4 of patients being positively effected. Percutaneous microballoon compression is safe for elderly patients.7 However, nearly all procedures cause some numbness and in a few this can be associated with intense pain obviating the whole point of the surgery ("anaesthesia dolorosa"). Microvascular decompression, although more risky, is increasingly used as it provides the longest period of pain relief and there is no sensory loss.
Complementary therapies
Due to the lack of curative measures the use of complementary therapies in trigeminal neuralgia has evolved quite rapidly. These include the following:TENS
Acupuncture
Biofeedback
Vitamin therapies e.g. vitamin B
Nutritional therapies e.g. garlic
However, there is no evidence available that support the use of these measures.
Prognosis
One third of patients will have mild symptoms and some will only ever have one episode. The pain of trigeminal neuralgia can be so intense it can lead to a poor quality of life due to mental and physical incapacity. Patients may require psychosocial input e.g. counselling. Untreated the condition worsens over time and although not fatal a patients life can be severely limited. The episodes become more frequent and more intense with time. At present there is a desperate need for further studies evaluating the role of current treatment modalities.
Clinical Context
A history of prior stroke is one of the most significant risk factors for stroke, and approximately one quarter of all strokes in the United States are recurrent. Moreover, transient ischemic attack is an important risk factor for stroke as well. The risk for stroke in the 90 days after a transient ischemic attack is 17%, with the greatest risk recorded during the first week after a transient ischemic attack.Medscape10/25/2010;
Fortunately, there are interventions available to patients and physicians to help prevent the risk for recurrent stroke. The current recommendations from the AHA and ASA provide an evidence-based guide to the prevention of recurrent stroke
Study Highlights
The risk for stroke begins to increase gradually when the systolic blood pressure increases past 115 mg Hg, and lowering blood pressure can reduce the risk for recurrent stroke by 30% to 40%. The ideal target blood pressure to prevent stroke has not been defined, but the authors suggest a level of 120/80 mm Hg or less.Similarly, the best regimen to reduce blood pressure among patients with a history of stroke is not completely clear, but diuretics, alone or in combination with angiotensin-converting enzyme inhibitors have been demonstrated to be useful agents.
More intensive treatment goals for diabetes are not necessarily beneficial in preventing recurrent stroke. Standard treatment goals for these patients should apply.
Statin therapy should be initiated to prevent recurrent stroke if the low-density lipoprotein cholesterol level is 100 mg/dL or if there is evidence of atherosclerosis. A treatment target for a low-density lipoprotein cholesterol level of less than 70 mg/dL is reasonable.
Niacin or gemfibrozil may be considered for patients with a low high-density lipoprotein cholesterol level.
Alcohol consumption should be limited to 2 drinks per day among men and 1 drink per day among women. Most evidence of any protective effect of alcohol against stroke is from primary prevention trials.
Although obesity can worsen the cardiovascular risk profile, it has not been directly linked with a higher risk for stroke. There are no trials that demonstrate that weight loss reduces the risk for recurrent stroke.
Patients who are able to exercise after stroke should perform at least 30 minutes of moderate-intensity physical activity at least 1 to 3 times per week.
There is insufficient evidence to recommend screening patients with a history of stroke for the metabolic syndrome.
Patients with 70% to 99% carotid stenosis with ipsilateral stroke or transient ischemic attack should be considered for carotid endarterectomy if the perioperative risk for morbidity and mortality is less than 6%. Carotid endarterectomy can be considered for certain high-risk candidates if the carotid occlusion is as low as 50%.
Carotid angioplasty and stenting may be considered as an alternative to carotid endarterectomy among patients at low to moderate risk for complications and who have carotid stenosis of more than 70% on noninvasive imaging.
For patients with stroke or transient ischemic attack from stenosis of a major intracranial artery, aspirin at a dose of 50 mg to 325 mg daily is preferred vs treatment with warfarin.
Warfarin should be used for patients with AF to prevent recurrent stroke, with aspirin reserved for patients who do not tolerate warfarin. "Bridge" therapy with low-molecular-weight heparin may be considered when warfarin treatment must be interrupted.
Conversely, the benefit of warfarin for secondary stroke prevention has not been proven among patients in normal sinus rhythm with a left ventricular ejection fraction of 35% or less.
Warfarin may be considered for patients with rheumatic mitral valve disease to prevent recurrent stroke.
Aspirin alone, aspirin plus extended-release dipyridamole, and clopidogrel may all be considered as first-line antiplatelet therapy to prevent recurrent stroke. The addition of extended-release dipyridamole to aspirin might help prevent approximately 1 additional stroke among 100 patients treated for 1 year.
Aspirin plus clopidogrel should not be used among these patients because of an elevated risk of bleeding.
There is no evidence in favor of increasing the dose of aspirin among patients who have a stroke while receiving aspirin therapy.
Postmenopausal hormone therapy is not recommended after stroke or transient ischemic attack.
New Metabolic Syndrome Recommendations
According to AHA/ASA criteria, metabolic syndrome is recognized when 3 of the following features are present:• increased waist circumference (≥102 cm in men; ≥88 cm in women),
• elevated triglyceride levels (≥150 mg/dL),
• reduced high-density lipoprotein cholesterol (<40 mg/dL in women; <50 mg/dL in men),
• elevated blood pressure (systolic ≥130 mm Hg, or diastolic ≥ 85 mm Hg), and
• elevated fasting glucose (≥100 mg/dL).
To prevent a secondary stroke or transient ischemic attack in patients who have metabolic syndrome, clinicians should treat the individual components of the syndrome that are also stroke risk factors, particularly dyslipidemia and hypertension, the new guidelines note.
Management of patients with metabolic syndrome should include counseling on diet, exercise, and weight loss to reduce vascular risks, but the utility of screening patients for metabolic syndrome after stroke has not been established.
There is considerable controversy surrounding this syndrome, largely because of uncertainty regarding its etiology and clinical usefulness, the authors write. The disorder has been related to an increased risk for diabetes, cardiovascular disease, and all-cause mortality.
The association between metabolic syndrome and risk for first ischemic stroke has been examined in several recent studies, with all except 1 confirming the association, but with only 1 study examining the association between metabolic syndrome and risk for stroke recurrence.
In the Warfarin Aspirin Symptomatic Intracranial Disease (WASID) trial, participants with metabolic syndrome were more likely to have a stroke, myocardial infarction, or vascular death during 1.8 years of follow-up than participants without metabolic syndrome (hazard ratio [HR], 1.6; 95% confidence interval [CI], 1.1 - 2.4; P = .0097). Patients with the syndrome were also at increased risk for ischemic stroke alone (HR, 1.7; 95% CI, 1.1 - 2.6; P = .012).
Cardiac features of metabolic syndrome improve with weight loss, which has also been shown to improve insulin sensitivity, lower plasma glucose, low plasma low-density lipoprotein cholesterol, lower plasma triglycerides, raise high-density lipoprotein cholesterol, lower blood pressure, reduce inflammation improve fibrinolysis, and improve endothelial function in patients with metabolic syndrome.
Neuropathic pain is often difficult to treat as it is resistant to many medications and effective medications often have adverse effects. Its estimated prevalence is between 1% and 2% in the United Kingdom.1 Treatment practice is thought to vary considerably throughout the UK in terms of starting treatment,
BMJ 2010;340
Neuropathic pain
achievement of therapeutic doses, and correct sequencing of therapeutic classes, thus probably leading in some cases to inadequate pain control, with considerable morbidity. This article summarises the most recent recommendations from the National Institute for Health and Clinical Excellence (NICE) on drug management for neuropathic pain in adults in primary and secondary care, excluding specialist pain services.
• First line treatment
• Offer oral amitriptyline or pregabalin as first line treatment (except for people with painful diabetic neuropathy):• -Amitriptyline: start at 10 mg a day, gradually titrating upwards to an effective dose or to the person’s maximum tolerated dose, with no dose higher than 75 mg a day .
• -Pregabalin: start at 150 mg a day (divided into two doses; a lower starting dose may be appropriate for some people), titrating upwards to an effective dose or to the person’s maximum tolerated dose, with no dose higher than a total 600 mg a day.
• If improvement is satisfactory, continue the treatment; consider gradually reducing the dose over time if improvement is sustained
• If amitriptyline as first line treatment reduces the pain satisfactorily but the person cannot tolerate the adverse effects, consider oral imipramine or nortriptyline as an alternative.
• Second line treatment
• If pain reduction is not satisfactory with first line treatment at the maximum tolerated dose, offer treatment with another drug type instead of or in combination with the original drug, after informed discussion with the person:• If the first line treatment was amitriptyline (or imipramine or nortriptyline), switch to or combine with oral pregabalin
• If the first line treatment was pregabalin, switch to or combine with oral amitriptyline (or imipramine or nortriptyline as an alternative if amitriptyline is effective but the person cannot tolerate the adverse effects).
For people with painful diabetic neuropathy:
If the first line treatment was duloxetine, switch to amitriptyline or pregabalin or combine with pregabalinIf the first line treatment was amitriptyline, switch to or combine with pregabalin.
• Third line treatment
• If pain reduction is not satisfactory with second line treatment, refer the person to a specialist pain service and/or a condition specific service, and while waiting for referral:• -Consider oral tramadol as a third line treatment instead of or in combination with the second line treatment; tramadol combined with amitriptyline, nortriptyline, imipramine or duloxetine is associated with only a low risk of serotonin syndrome (features of which include confusion, delirium, shivering, sweating, changes in blood pressure, and myoclonus)
Consider topical lidocaine for localised pain in people unable to take oral medication because of medical conditions and/or disability; topical lidocaine is licensed for post-herpetic neuralgia but not for other neuropathic pain.
For tramadol as monotherapy, start at 50-100 mg not more often than every four hours, titrating upwards if required to an effective dose or to the person’s maximum tolerated dose, with no dose higher than 400 mg a day. If tramadol is used as combination therapy, more conservative titration may be required.
Other treatments
Do not start treatment with opioids (such as morphine or oxycodone) other than tramadol without an assessment by a specialist pain service or a condition specific service.Drug treatments other than those recommended in this guideline that are started by a specialist pain service or a condition specific service may continue to be prescribed in non-specialist settings, with a multidisciplinary care plan, local shared care agreements, and careful management of adverse effects.
• Summary points
• Huntington’s disease causes motor, cognitive, and psychiatric impairment• Predictive and diagnostic genetic testing are available through specialist centres
• Genetic testing for the disease has many implications for patients and families
• The disease currently has no cure, but many therapeutic options exist to improve symptoms
• Optimal care usually requires input from a multidisciplinary team
BMJ 2010;340
Huntington’s disease
• Common symptoms of Huntington’s disease
• Motor symptoms• Chorea, dystonia, loss of postural reflexes, bradykinesia, rigidity
• Cognitive symptoms
• Disorganisation as a result of difficulties with planning, initiating, and organising thoughts, activities, and communication; perseveration; impulsivity; perceptual distortions; lack of insight; distractibility; difficulty in learning new information
Psychiatric
Depression, obsessive-compulsive disorders, anxiety, irritability, apathy, hypersexuality (uncommon), psychosis (uncommon)Metabolic
Weight loss, sleep disturbance
Others
Dysphasia (combination of motor and language difficulties), dysphagia (combination of motor problems, impulsivity, and distractibility)
Motor symptomsThe motor symptoms of Huntington’s disease can be divided into two categories: added involuntary movements such as chorea and impaired voluntary movements, which cause limb incoordination and impaired hand function. These symptoms are worsened by loss of postural reflexes. The pattern of symptoms tends to change over time, with chorea declining and dystonia, rigidity, and bradykinesia becoming more marked.
Cognitive symptomsCognitive impairment includes slowing of thought processing and deterioration of executive functions (high level cognitive processes that control other aspects of cognitive function). Typically, patients report difficulty with multitasking, concentration, and short term memory. Thinking style becomes more concrete and less efficient, and the planning, initiation, and organisation of time, thoughts, and activities become harder. People withHuntington’s disease are often impulsive and develop psychomotor perseveration. Visuospatial perception can also deteriorate.
Psychiatric symptomsDepression is one of the most common psychiatric symptoms and occurs as part of the disease, rather than merely as a response to diagnosis. A recent survey of 2835 patients with the disease found that 40% had symptoms of depression, and 50% reported having sought treatment for depression in the past. Other reported psychiatric symptoms include obsessive-compulsive symptoms and psychosis.
It is important to recognise psychiatric symptoms in Huntington’s disease so that symptomatic treatment can be offered. This may be difficult later in the disease because diagnoses may be obscured by other features of the disease; depression, for example, may be difficult to detect in a patient who has altered facial expressions and tone of voice. Conversely, metabolic symptoms such as weight loss and sleep disturbance may be wrongly attributed to depression.
Suicide riskPatients with Huntington’s disease are more likely than members of the general population to commit suicide according to a meta-analysis of studies that reported mortality associated with mental disorders (standardised mortality ratio of 290).w6 A survey of 4171 carriers of the Huntington’s gene with premanifest and manifest disease found that 17.5% had suicidal thoughts at or around the time of assessment and 10% of those surveyed had made at least one suicide attempt in the past.11
Suicidal ideation was highest in gene carriers who were nearing the threshold of being diagnosed with manifest disease (those with soft motor signs of Huntington’s disease), and in those who were beginning to lose their functional ability and independence (those with stage 2 disease). Risk factors for suicide in Huntington’s disease include depression and impulsivity.4 Some people with the disease also have suicidal thoughts in the absence of depressionw7: for some, thoughts of suicide seem to be a rational response to their imminent loss of independence.
Metabolic symptomsHuntington’s disease causes metabolic symptoms, which include catabolic weight loss, endocrine dysfunction, and sleep disturbance.12
Advanced diseaseBy the time patients have endstage disease they are profoundly disabled. Communication may be severely limited and muteness is common, which can result in agitation and frustration. Huntington’s disease does not cause global dementia, however, and the ability to recognise and interact with people is often preserved. Huntington’s disease is a catabolic condition, and this, combined with marked dysphagia, means that it can be difficult to provide sufficient nutrition to maintain a patient’s weight.
How is Huntington’s disease inherited?
What is the genetic basis of the disease?Huntington’s disease is a single gene disease with autosomal dominant inheritance. The genetic abnormality is an expanded CAG trinucleotide repeat within the Huntingtin (HTT) gene on chromosome 4, and it can be identified through genetic testing.13 The HTT gene encodes the protein huntingtin, which is essential for normal neural development, although its functions are incompletely understood.14 w8 w9 In Huntington’s disease, the expanded HTT gene encodes a mutant form of huntingtin protein.The mutant protein causes or contributes to the development of Huntington’s disease through many pathogenic mechanisms.
Offspring of an affected parent have a 50% chance of inheriting the genetic abnormality, and males and females are affected equally. Huntington’s disease does not skip generations.
What is the meaning of CAG repeat length in Huntington’s disease?A "normal" HTT gene has fewer than 36 repeats. The gene is abnormal, or expanded, if it has 36 or more repeats, and CAG repeats of 40 or more will always cause Huntington’s disease. Genes with CAG repeat lengths between 36 and 39 show reduced penetrance, which means that some people with these lengths will develop Huntington’s disease and some will not; those who do develop disease are likely to develop later onset disease.w10 An intermediate repeat length between 29 and 35 does not cause the disease but may expand into the pathogenic range in future generations.
The instability of intermediate alleles is one cause of sporadic Huntington’s disease, in which the disease develops in someone with no apparent family history. Apparent sporadic Huntington’s disease occurs in 6-8% of new cases of Huntington’s disease,w11 w12 and it can also be caused by unexpected or unknown paternity, or a parent dying before they develop symptoms of the disease.
Instability of the CAG repeats can also cause "genetic anticipation," in which the CAG repeat length increases and causes onset of the disease at a younger age in affected offspring than in the parent. Genetic anticipation is more common when the expanded allele is inherited from a father than from a mother. About 90% of people with juvenile disease (with CAG repeats typically >60) inherit the mutation from their father.w13 w14
CAG repeat length is related to age of onset of disease at a population level: the longer the CAG repeat length, the earlier the onset of symptoms tends to be. However, repeat length only accounts for 50-70% of this variance, and disease onset in an individual cannot be predicted reliably through genetic testing.w15 The CAG repeat length is related less strongly to the rate of disease progression.
How is genetic testing undertaken?
Genetic testing for Huntington’s disease is performed by measuring the CAG repeat length in the HTT gene. We will use the term "positive" test result to refer to CAG lengths in the pathogenic fully penetrant CAG repeat range of more than 39 repeats. Testing falls into two categories.Diagnostic testing is carried out to confirm (or refute) the diagnosis in a patient with symptoms suggestive of Huntington’s disease. It is a test for manifest disease and is most commonly undertaken by neurologists. A positive diagnosis has numerous implications for family members (especially children and siblings) and for the patient, and it is important to offer as much information as possible about the disease and the meaning of a positivediagnosis before testing. Our experience has been that family members’ reactions and coping strategies are linked to how they find out that they are at risk themselves.
Predictive testing is carried out in a person who has no symptoms of the disease, but who is at risk because of their family history. It determines whether that person carries the expanded HTT gene and will develop Huntington’s disease in the future. A positive predictive test result indicates that they will certainly develop Huntington’s disease at some point in the future (unless they die of another cause in the meantime), but it does not tell them when this will happen or what the presenting symptoms will be.
Reasons commonly cited for having predictive testing include wishing to relieve uncertainty, to inform decisions about reproduction, and to plan for the future. Predictive testing is not performed in children. This is because informed consent requires an adult understanding of the consequences of a predictive test result. Consent for predictive testing by a parent would remove from a child their right to autonomy in choosing whether or not to be given this information.
Predictive testing for Huntington’s disease is performed in specialist genetic centres and follows internationally agreed guidelines. These include an initial session of pre-test counselling, followed by a period of reflection, and then a second session of counselling. Post-test counselling must also be available. Matters such as insurance and mortgages are discussed (see below).
Several studies have reviewed the uptake of predictive testing among people at risk; on average, 5-20% of those at risk in the United Kingdom take up the option.
• What are the implications of a positive gene test for the individual?
• Emotional implicationsA patient who receives a diagnosis of manifest Huntington’s disease learns that he or she has a slowly progressive incurable disease. A person who receives a positive predictive test experiences the extra burden of being uncertain when the disease will begin to take effect. Most people who have genetic testing for Huntington’s disease will have watched a parent develop the disease, will be familiar with the change in personality and cognition that it causes, and will know what effect their illness will have on those around them.Since direct genetic testing for Huntington’s disease became available in 1993, a steadily increasing cohort of premanifest gene carriers has been created. Many of this group may not wish to engage with health or social services until they develop manifest disease, whereas others value emotional and practical support to help them deal with the implications of a predictive test result.
In 1996, one study followed up 135 Canadians who had entered a predictive testing programme.w21 Adverse events, including psychological distress, substance misuse, and relationship breakdowns, were recorded in seven of the 37 who received a positive result compared with eight of the 58 who received a negative result. (The remaining subjects received equivocal results or chose not to proceed with testing.) One subject in each group reported suicidal ideation.
A positive predictive test can come as a relief, however, because some people find a positive test result easier to cope with than the uncertainty of being at risk. As discussed above, only a minority of people at risk choose to have predictive testing; these people are a self selecting group, and the aim of genetic counselling is to ensure that they are equipped to deal with their predictive test result. Diagnostic testing can also be helpful in removing uncertainty—for example, a diagnosis of Huntington’s disease could alleviate the distress caused by an unexplained change in personality.
Huntington’s disease is, however, the one disease that is exempt from a total ban on disclosure; applicants must disclose Huntington’s disease predictive test results if they apply for life insurance over £500 000. A negative predictive test result will bring insurance premiums back in line with those of people without a family history of Huntington’s disease. This insurance moratorium is due to be reviewed in 2011.
A positive predictive test does not need to be declared when applying for travel insurance in the UK, but manifest disease does. Specialist companies with experience of insuring people with pre-existing medical conditions are helpful, and the HDA can provide details of appropriate contacts.
Under the 1995 Disability Discrimination Act in the UK, it is illegal for employers to discriminate against someone who is disabled by dismissing them or by treating them negatively because of their disability.
We strongly recommend that patients with Huntington’s disease who feel that their ability to work is deteriorating inform their employers about their diagnosis to ensure that their job is legally protected. Once diagnosis is revealed, regular assessment should take place according to occupational risk.
The legal situation for people at risk of Huntington’s disease because of their family history is less clear cut, and there is currently no law in the UK to prevent discrimination against those with a genetic diagnosis (in employment, or elsewhere). A working group of the Human Genetics Commission in the UK is currently examining this area and is due to report in 2011. In the United States, the 2008 Genetic Information Nondiscrimination Act prohibits discrimination on the basis of genetic information in employment and health insurance provision.
In the UK, asymptomatic gene carriers are not obliged to disclose a positive predictive test result to their employer unless a health questionnaire has a specific question on this. In reality, however, some employers may place restrictions, based on family history, on the work of people who are at risk or are asymptomatic gene carriers. The police, for example, have separate rules for each authority but may ask employees at risk to have regularneurological reviews.
UK armed forces are a special case: until recently, applicants to the armed forces who were at risk of Huntington’s disease because of their family history were accepted only if they had received a negative predictive test result. This policy is now being reconsidered, with alternatives such as a shorter term of service being offered. As with patients with manifest Huntington’s disease, we recommend regular review of premanifest gene carriers as appropriate for their occupational level of risk.
Research on genetic discrimination in Huntington’s disease is limited, but a recent survey of 233 tested and untested people at risk of Huntington’s disease in Canada found that 6.4% reported genetic discrimination related to employment.w22 Anecdotal evidence also suggests that some premanifest gene carriers have been discriminated against in the UK.
What are the implications of a positive Huntington’s disease gene test for the family?
A positive predictive or diagnostic Huntington’s disease test has a huge effect on the partner and family of the person tested. Someone close to the person tested faces the prospect of becoming a carer, often when relatively young, and probably for many years. A new diagnosis of Huntington’s disease has implications for siblings and children who suddenly become at risk of acquiring the disease.
Rarely, an at risk person’s positive test result will in effect tell another family member their own gene status. For example, if the grandchild of someone with Huntington’s disease, who will have a 25% risk of carrying the Huntington’s disease gene, has a positive test, their at risk parent must be a gene carrier.
If the intermediate family member does not wish to know their gene status, every effort should be made to find a solution that is acceptable for all parties. This situation is rare, but guidelines state that if no agreement is reached after all attempts for an acceptable compromise, the wishes of the person at 25% risk should be followed and testing performed.
The dynamics of families affected by Huntington’s disease are often complicated, particularly between those who do and those who do not carry the expanded gene. It may create supportive and close knit families, but it can also be extremely divisive. The result is often a high level of resilience and care within the family, mixed with anger, resentment, and guilt.
Anger and resentment can have many outlets: towards a parent who has passed on their expanded gene; towards unaffected family members, particularly siblings; or towards older generations who have not been open about the presence of Huntington’s disease in the family. Guilt also has many manifestations, such as that felt by a parent who has passed on their expanded gene to a child, or who has become symptomatic and now sees himself or herself as a burden to the family, or survivor guilt in people who have had a negative predictive test.
Receiving a negative test result when other family members have tested positive can be an isolating experience too.
Most affected families will contain more than one person with the disease, with several more at risk of developing the disease in the future, creating a scenario where one person first endures the burden of being a carer and being bereaved by the disease, and later being a patient who needs care.
How is Huntington’s disease managed?
The aim of treatment is to manage symptoms and improve quality of life. No current treatments can slow disease progression, although promising disease modifying treatments are being tested preclinically in animal models of Huntington’s disease.15 22There are many effective options for symptomatic management, however, both drug based and non-drug based.23 24 Tables 1 and 2 summarise those drugs commonly used for symptom management. Their choice is based mainly on clinical experience because the evidence base for drugs in Huntington’s disease is small.25 26 27 28 Tetrabenazine has the best evidence of efficacy in Huntington’s disease and has been shown to reduce chorea in a randomised controlled clinical trial.w23 Box 2 contains details of where to access further information about drug treatments in Huntington’s disease.
• What ethical considerations surround genetic testing?
• Confidentiality and consentAccording to internationally agreed guidelines, strict confidentiality is observed before, during, and after predictive testing. No one other than the adult being tested is told about the process without their express permission. This includes partners and family members.Some people with positive predictive Huntington’s disease test results choose not to inform their general practitioner so that the information remains confidential.Written informed consent must be obtained from the patient before either predictive testing or diagnostic testing. If the patient lacks capacity to make the decision themselves for diagnostic testing, consent can be given by an authorised representative.
Having childrenDeciding whether or not to have children is often difficult for people with or at risk of having an expanded HTT gene. A minority choose to have either prenatal testing or preimplantation genetic diagnosis, which ensures that their child has a less than 1% chance of carrying the mutant gene. Although couples in which one partner is at risk of or has Huntington’s disease are not usually eligible to adopt, some adoption agencies will consider it, and fostering may also be possible.
Prenatal testingPrenatal testing is usually carried out via chorionic villus sampling between 11 and 13 weeks of pregnancy. Pre-test counselling is important: potential parents need to be sure that they will terminate the pregnancy if their fetus is found to have an expanded HTT gene, otherwise their child will grow up in the shadow of a predictive test for which they did not consent. This would violate the autonomy of the child. A termination cannot, of course, be forced on a couple who change their mind after receiving a positive test result.
These considerations, together with the potential risk of miscarriage after chorionic villus sampling or amniocentesis, mean that prenatal testing cannot be undertaken lightly. In practice, a couple who disclose a family history of Huntington’s disease for the first time in pregnancy only rarely proceed to prenatal testing.
Preimplantation genetic diagnosis This technique is available through specialist units. In preimplantation genetic diagnosis, embryos are created using normal in vitro fertilisation procedures and then tested for the expanded HTT gene. Unaffected embryos are implanted. Overall, about one in five cycles results in a live birth, but success rates vary. The Human Fertilisation and Embryology Association website contains more details (www.hfea.gov.uk/).
Exclusion (non-disclosing) prenatal testing and preimplantation genetic diagnosisExclusion (non-disclosing) prenatal testing or preimplantation genetic diagnosis can be carried out for couples in which one partner is at risk but does not wish to have a predictive test. With this test the potential parents do not find out their own Huntington’s disease gene status. The test requires blood samples from several family members, and linkage techniques are used rather than testing for the CAG expansion (see fig 2 for more details).
A "high risk" result means that the fetus is at 50% risk of developing Huntington’s disease—the same risk as the at risk parent. A couple undergoing this test may choose to terminate a pregnancy at 50% risk. Clearly this test requires detailed discussion with the couple beforehand. In practice, very few of these tests are performed, but it is a helpful option for some couples.
Managing the movement disorderThe first step is to decide whether symptoms need treating. Patients are often not bothered by early chorea, for example, and may not even be aware of it. As chorea develops, however, it can interfere with voluntary activities like writing or eating, and it often causes falls, making intervention necessary. Chorea can be distressing in itself, and patients often find themselves accused of drunkenness by people unaware of their diagnosis.
Non-drug interventions should be considered first. Chorea often varies with posture or positioning, and devices such as padded chairs or wrist and ankle weights to reduce the amplitude of chorea may be helpful. Shoes with non-slip soles and grab rails around the home can improve safety, and assessment of the home by an occupational therapist is often extremely useful.
Physiotherapy can also help optimise mobility, and can be especially beneficial early in the disease to preserve mobility and independence for as long as possible. Like most involuntary movements, chorea is worsened by stress, anxiety, and depression, so treating these and providing a calm predictable environment are beneficial.
If these measures are not sufficient to control symptoms, drugs can be tried. These are unlikely to prevent chorea completely, but they can dampen down symptoms considerably, and tetrabenazine is generally the first choice.
It can exacerbate or trigger psychiatric symptoms, so it should be avoided in patients with a history of depression or other psychiatric disorders, and in these patients, or in those in whom symptoms are not controlledwith tetrabenazine, the atypical neuroleptics are helpful. Of these, olanzapine is our first choice because it is an effective anti-chorea drug that also promotes weight gain, suppresses irritability, and improves sleep—all of which are useful side effects in Huntington’s disease.
Movement suppressing drugs used in the earlier stages may however exacerbate the impaired movements that develop later on. They will often need to be reduced and eventually stopped, so regular reassessment is vital.
Managing cognitive and psychiatric manifestationsRecognising and acknowledging that these symptoms are probably caused by Huntington’s disease is important so that strategies can be developed to deal with them. It is important to ask depressed patients about suicidal thoughts, and to take a proactive approach to the treatment of psychiatric symptoms. Treatment of depression in Huntington’s disease is with standard antidepressants. Although there is not an established evidence base for the treatment of depression in Huntington’s disease, our experience is that antidepressants are often very effective.
Psychological treatments, such as cognitive behavioural therapy, can also be helpful in selected patients. This can also be a useful way for people with premanifest disease to learn cognitive strategies that will stand them in good stead once they develop cognitive and psychiatric symptoms. We know of no controlled trials of this treatment in Huntington’s disease, although one case study reported benefit for a patient with premanifest disease.w24 Support from local community mental health teams is often invaluable.
No drug treatments are available for cognitive symptoms, but coping strategies can often be adopted to overcome problems or compensate for them. Difficulty with multitasking is an example of a common early symptom; concentrating in a busy office can be difficult and tension may develop in relationships with colleagues.
This, in turn, will increase stress and exacerbate the underlying problem with concentration. Once these problems have been identified, appropriate compensatory strategies can be devised, such as moving into a quiet office and reducing workload. Employers have a statutory duty to optimise the working environment for people with a disability where possible, although this needs to be approached sensitively.
Apathy is also a challenging symptom to manage and can be difficult to differentiate from depression; it may be worth considering a trial of antidepressants if this is the case. It is important to gently impose structure on the day, because patients often find that having an appointment to aim for, such as coffee with a friend, helps them to initiate activities and organise their behaviour. It can be difficult for people with Huntington’s disease to initiate activities, but once started they are often able to participate fully with encouragement and support from carers.
End of life carePlanning for end of life care raises several ethical problems that often relate to how far medical interventions should be taken in patients who no longer have the capacity to make their wishes known. We find that advanced decisions to refuse treatment (previously known as advanced directives) can be extremely helpful.
They give patients the security of knowing that their wishes will be carried out, even if they are no longer able to makedecisions or communicate, and they relieve relatives of the responsibility of making choices. Advanced decisions to refuse treatment and end of life care should be raised as early as possible so that they can be discussed by the patient and their loved ones.
As Huntington’s disease progresses, it often becomes increasingly difficult to provide care at home, and a nursing home may be the best option. Insertion of a gastrostomy tube may be appropriate in patients who are unable to maintain adequate nutrition and body weight. Box 3 contains sources of information for palliative management in Huntington’s disease.
Conclusions
Managing the many facets of Huntington’s disease can be challenging and is best served within multidisciplinary settings. We continue to learn about how to improve our services from our patients and their families. The current major push to find disease modifying treatments, coupled with the existence of a specific and sensitive diagnostic genetic test, means that in the future treatments might be initiated in the premanifest phase, with the hope of delaying or halting the disease process itself.• Current research
• There is currently a major push to find disease modifying drugs and new treatments for the symptoms of Huntington’s disease, and many new developments have been made in recent years
• In February 2010, the results of the MermaiHD study were announced as a press release: pridopidine, a dopamine stabiliser (also known as Huntexil, previously known as ACR16) was shown to improve voluntary motor symptoms in a phase III trial of 437 patients with Huntington’s disease. Publication in a peer reviewed form is awaited
• Previous clinical trials have evaluated creatine and coenzyme Q10 among other compounds, but have not demonstrated efficacy. Many patients continue to buy them over the counter and comparison trials of the two substances, funded by the National Institutes for Health, are ongoing
BMJ 2010;340
A phrase III trial of latrepirdine (Dimebon), is currently taking place in multiple sites, including several in the UK
Exciting progress has been made recently in identifying several other pathways that are potential targets for future therapeutic intervention and clinic trials. This is too large a subject to cover in detail, but see Imarisio et al for a comprehensive review. Potential therapeutic approaches include:
Enhancing clearance of mutant huntingtin by cellular clearance mechanisms: several compounds being tested in mouse models of Huntington’s disease aim to promote clearance of the mutant protein, huntingtin, which is generated by the expanded HTT gene
Histone deacetylase inhibitors: these target the transcriptional dysregulation that occurs early in the pathogenesis of Huntington’s disease
Inhibitors of proteolytic cleavage of full length mutant huntingtin: these would prevent production of the potentially toxic N-terminal fragment
Gene silencing: to switch off expression of the mutant gene itself
To achieve optimal effect, future disease modifying treatments will probably consist of a combination of treatments targeting key pathogenic pathways, similar to the treatment of HIV or cancer
Much progress has been made in developing and evaluating sensitive biomarkers that will help to measure the effects of disease modifying treatments in future clinical trials, particularly in the premanifest and early stages of the disease. Track-HD7 and Predict-HD9 are major international collaborative studies that have increased our understanding of the course of Huntington’s disease and which biomarkers are the best for monitoring this
Diagnosis, prevention, and management of delirium: summary of NICE guidance
BMJ 2010;341• Delirium is a complex clinical syndrome characterised by disturbed consciousness, cognitive function, or perception. Sometimes known as acute confusional state, delirium has an acute onset, a fluctuating course, and is associated with serious adverse outcomes such as death, dementia, and the need for long term care.
Although common in general hospitals (affecting as many as about 30% of inpatients)1 and care homes, delirium is oftenpoorly recognised1; however, it can be prevented in about one third of patients at risk. This article summarises the most recent recommendations from the National Institute for Health and Clinical Excellence (NICE) guideline on how to recognise, prevent, and treat delirium.
Recommendations
NICE recommendations are based on systematic reviews of best available evidence and explicit consideration of cost effectiveness. When minimal evidence is available, recommendations are based on the experience of the Guideline Development Group and their opinion of what constitutes good practice. Evidence levels for the recommendations are given in italic in square brackets.Assessment of risk factors
When people first present to hospital or long term care, assess them for the presence of the following risk factors for delirium:• -Age 65 years or older
• -Cognitive impairment (past or present), dementia, or both. If cognitive impairment is suspected, confirm it using a standardised and validated cognitive impairment measure, such as the mini mental state examination
• -Current hip fracture
• -Severe illness (that is, a clinical condition that is deteriorating or at risk of deterioration).
• Interventions to prevent delirium
• Ensure that people at risk of delirium are cared for by a team of healthcare professionals who are familiar to the person at risk (that is, do not change staff excessively during the person’s stay in hospital or long term care).• Avoid moving people within and between wards or rooms unless absolutely necessary.
• [Both the above recommendations are based on low quality evidence from a prospective cohort study and the experience and opinion of the GDG]
Within 24 hours of admission, assess people at risk for the following clinical factors that might precipitate delirium:
• -Cognitive impairment, disorientation, or both
• -Dehydration, constipation, or both
• -Hypoxia
• -Immobility or limited mobility
• -Infection
• -Multiple medications
• -Pain
• -Poor nutrition
• -Sensory impairment
• -Sleep disturbance.
Indicators of deliriumAssess for recent (within hours or days) changes or fluctuations in behaviour. Assessment should be done at presentation for all people at risk and, subsequently, at least daily for all people admitted to hospital or long term care. These behaviour changes may be reported by the person at risk, or by a carer or relative, and may affect:
Cognitive function—for example, worsened concentration*; slow responses*; confusion
Perception—for example, visual or auditory hallucinations
Physical function—for example, reduced mobility*; reduced movement;* restlessness; agitation; changes in appetite*; sleep disturbanceSocial behaviour—for example, lack of cooperation with reasonable requests; withdrawal*; or alterations in communication, mood, or attitude.
Be particularly vigilant for behaviour changes that suggest hypoactive delirium (marked *). [Based on the experience and opinion of the GDG]
Diagnosis (by specialist clinical assessment)
If indicators of delirium are identified, carry out a clinical assessment based on the criteria in the Diagnostic and Statistical Manual of Mental Disorders, fourth edition, or the short confusion assessment method (CAM) to confirm the diagnosis. In critical care or in the recovery room after surgery, the confusion assessment method for the intensive care unit (CAM-ICU should be used.A healthcare professional who is trained and competent in the diagnosis of delirium should carry out the assessment. If distinguishing between the diagnoses of delirium, dementia, and delirium superimposed on dementia proves difficult, treat for delirium first.
Ensure that the diagnosis of delirium is documented both in the person’s hospital record and in his or her primary care record.
Initial management of people with delirium
Identify and manage the possible underlying cause(s). [Based on moderate and low quality evidence from randomised and non-randomised studies and the experience and opinion of the GDG]Ensure effective communication and reorientation (for example, explain where the person is, who they are, and what your role is) and provide reassurance. Consider involving family, friends, and carers to help with this. [Based on evidence from a low quality prospective cohort study, qualitative studies, and the experience and opinion of the GDG]
Provide a suitable care environment. (Please refer to the first two points in the section entitled "Interventions to prevent delirium.") [Based on evidence from a low quality prospective cohort study and the experience and opinion of the GDG]
Distressed people
If a person with delirium is distressed or considered a risk to themselves or others, use verbal and non-verbal de-escalation techniques.8 If these are ineffective or inappropriate, consider giving short term (usually one week or less) haloperidol or olanzapine. Start at the lowest clinically appropriate dose and titrate cautiously according to symptoms. [Based on low quality evidence from a randomised trial and the experience and opinion of the GDG]
BMJ 2010;341
Use antipsychotic drugs with caution or not at all for people with conditions such as Parkinson’s diseaseor dementia with Lewy bodies because such treatment increases the risk of harms such as increased extrapyramidal symptoms and the neuroleptic malignant syndrome. [Based on the experience and opinion of the GDG]
Overcoming barriers
Effective implementation of these recommendations will require three inter-related actions from health professionals, care staff, and their employing organisations. Firstly, awareness of delirium as a common and serious illness ("think delirium") should be improved. The current poor detection of delirium1 should be addressed by education programmes.Use existing clinical codes to document the condition for audits and as an indicator of care quality and outcomes.
Secondly, staff in hospitals and care homes should adopt a new culture of preventing delirium. This will require: specific education to change attitudes and improve knowledge and skills; a re-design of care systems; and clear clinical and managerial leadership and responsibility, because a whole hospital or a whole care home approach is necessary.
Lastly, greater involvement of patients and relatives is required to inform timely patient centred care (the key to delirium prevention) and the redesign of care systems (for example, ward noise reduction, maintaining hydration and nutrition).
• Summary points
• The vegetative state is a complex neurological condition in which patients appear to be awake but show no sign of awareness of themselves or their environment• Current clinical methods of diagnosis are limited in scope, evidenced by a high rate (about 40%) of misdiagnosis (that is, patients who are aware are considered to be unconscious)
The vegetative state
BMJ 2 August 2010;341
The main causes of misdiagnosis are associated with a patient’s disability (such as blindness), confusion in terminology, and lack of experience of this relatively rare condition
Furthermore, standard behavioural assessments cannot distinguish an aware (that is, minimally conscious) but completely immobile patient from a non-aware patient (one with vegetative state).
In such behaviourally non-responsive patients, functional neuroimaging methods (such as magnetic resonance imaging or electroencephalography) can detect residual cognition and awareness and can even establish two way communication, without requiring any behavioural output from patients
Current guidelines should therefore be modified to include functional neuroimaging as an independent source of diagnostically relevant information
• The vegetative state may develop suddenly (as a consequence of traumatic or non-traumatic brain injury, such as hypoxia or anoxia; infection; or haemorrhage) or gradually (in the course of a neurodegenerative disorder, such as Alzheimer’s disease).
• Although uncommon, the condition is perplexing because there is an apparent dissociation between the two cardinal elements of consciousness: awareness and wakefulness.
Patients in avegetative state appear to be awake but lack any sign of awareness of themselves or their environment.w1 Large retrospective clinical audits have shown that as many as 40% of patients with a diagnosis of vegetative state may in fact retain some level of consciousness.
Misdiagnosis has many implications for a patient’s care—such as day to day management, access to early interventions, and quality of life—and has ethical and legal ramifications pertaining to decisions on the discontinuation of life supporting therapies.
Overall, our understanding of the vegetative state is incomplete. Although we know quite a lot about the neuropathology underlying the vegetative state, our ability to assess (un)consciousness and cognitive function in the clinic is extremely limited, as highlighted by the high rate of misdiagnosis.
What is the vegetative state and what is it not?
The 2003 guidance from the UK’s Royal College of Physicians on diagnosing and managing the permanent vegetative state defines it as"a clinical condition of unawareness of self and environment in which the patient breathes spontaneously, has a stable circulation, and shows cycles of eye closure and opening which may simulate sleep and waking."
Three main clinical features define the vegetative state:
(a) cycles of eye opening and closing, giving the appearance of sleep-wake cycles (whether the presence of eye opening and closing cycles actually reflects the presence of circadian rhythms is unclear);(b) complete lack of awareness of the self or the environment; and
(c) complete or partial preservation of hypothalamic and brain stem autonomic functions.
Although both the persistent and the permanent vegetative states are often abbreviated to "PVS," authors of a letter in the BMJ in 2000 suggested that to avoid confusion the abbreviation should be used exclusively to indicate a permanent vegetative state.w7 The American Congress of Rehabilitation Medicine suggested that the cause of injury (traumatic, anoxic) as well as the time elapsed since onset of the condition should be documented, as both are important for prognosis.
Experts have suggested that the vegetative state should be seen as part of a continuous spectrum of conditions, often referred to as disorders of consciousness, in which someone’s wakefulness and/or awareness are impaired after severe brain injury (figure , table 1 ). This suggestion is consistent with the idea that awareness and unawareness are part of a continuum, and it highlights the importance of differentiating the vegetative state from other related neurological conditions that may also follow catastrophic brain injury.
• Coma Coma is a condition of unresponsiveness in which patients lie with their eyes closed, do not respond to attempts to arouse them, and show no evidence of awareness of self or of their surroundings.
• Patients lack not only signs of awareness (similar to vegetative state) but also wakefulness (unlike vegetative state) regardless of how intensely they are stimulated. Patients typically either recover or progress to a vegetative state (that is, they show signs of wakefulness) within four weeks. Irreversible coma with absent brainstem reflexes indicates brain death, which is not the same as a vegetative state.
Minimally conscious stateThe minimally conscious state is a condition in which patients appear not only to be wakeful (like vegetative state patients) but also to exhibit inconsistent (fluctuating) but reproducible signs of awareness (unlike patients with vegetative state). Like the vegetative state, the minimally conscious state may be transitory and precede recovery of communicative function or may last indefinitely.
Locked-in syndromeLocked-in syndrome (or pseudocoma), although not a disorder of consciousness, may be confused with vegetative state. Patients with locked-in syndrome are both awake and aware, yet they are entirely unable to produce any motor output or they have an extremely limited repertoire of behaviours (usually vertical eye movement or blinking).
What causes the vegetative state?
In terms of neuropathology, the vegetative state is mostly marked by cortical or white matter and thalamic, rather than brain stem, injury. A review of the evidence available up until 1994 highlighted the fact that traumatic injury was found to be associated with diffuse damage to subcortical white matter (or diffuse axonal injury).Cases of non-traumatic injury, on the other hand, were found to have extensive necrosis in the cerebral cortex, almost always associated with thalamic damage.
In a more recent survey of patients with brain injury (n=49), 35 (71%) patients had traumatic brain injury, of whom 25 (71%) had severe diffuse axonal injury and 7 (20%) had major injury to the cerebral cortex.
Among the 35 patients, the thalamus seemed to be abnormal in 28 (80%) and damage to the brain stem was present in only 5 (14%). In the 14 (29%) patients with non-traumatic injury, 9 (64%) cases presented with diffuse neocortical damage; in all 14 cases a profound and diffuse neuronal loss was apparent in the thalamus and hippocampus. Overall, these lesions effectively render a structurally intact cortex unable to function by destroying the connections between cortical areas via the thalamus, as well as afferent and efferent cerebral connections.
What affects prognosis in patients with a diagnosis of vegetative state?
Three major factors affect the prognosis of patients with vegetative state:• time spent in the vegetative state,
• age, and
• type of brain injury.
Time spent in the vegetative stateA study of 140 patients showed that time spent in a vegetative state is negatively correlated with the chances of recovering independence and consciousness and positively correlated with the probability of remaining in a vegetative state.12 The role of time in prognosis was confirmed by a large review of 603 adult published cases,13 from which it was estimated that the chance of regaining independence at one year after injury steadilydecreased with time from 18% (one month in the vegetative state), to 12% (three months), and 3% (six months).
Similarly, the chance of recovering consciousness at one year also decreased, from 42% to 27% and 12% respectively. The chances of remaining in the vegetative state at one year after injury were estimated to increase from 19% to 35% and 57% respectively.
Although several protocols exist for conducting behavioural assessments (articles by Giacino et al and Majerus et al provide an overvieww15 w16), they differ greatly in their ability to detect consciousness because of the number of domains (such as arousal and vision) assessed and the thoroughness of the assessment. Indeed, a recent study of 60 patients compared on three assessment techniques reported that the Glasgow coma scalew17classified as vegetative several patients who showed signs of consciousness according to other behavioural scales.15
The Full Outline of UnResponsiveness (FOUR) reclassified 13% of the supposedly vegetative patients as minimally conscious, and the coma recovery scale-revised (CRS-R)w19 reclassified an additional 28% of the patients as minimally conscious. The main discrepancy between scales seems to relate to their different focus on oculomotor behaviour, with the FOUR and CRS-R protocols testing a greater variety of visual behaviours. For example, in all the patients reclassified by the CRS-R protocol, visual fixation was the key behaviour indicating awareness.
Does misdiagnosis of the vegetative state occur?
According to accumulating evidence from retrospective clinical audits and comparisons of alternative behavioural assessment techniques, misdiagnosis of minimally conscious patients as being in a vegetative state is not uncommon. In particular, although some studies have reported relatively low rates of misdiagnosis (18% ), most studies seem to converge, across time and geographical location, on an approximate rate in excess of 40% .Errors in diagnosis may result from lack of skill or training in the assessment of patients with catastrophic brain injury, limited knowledge of this relatively rare condition, and confusion in terminology.
Two main problems seem to underlie misdiagnosis.
Firstly, behavioural assessments of awareness present many complexities. For example, patients with physical disability may not be able to respond to stimulation—something that was true in all misdiagnosed cases in a large retrospective study of 97 patients with profound brain damage.2 Sensory impairments (particularly in the visual domain) can also mask the presence of awareness,16 20 a factor that has been reported as underlying as many as 65% of misdiagnoses.2Other acquired conditions, such as hydrocephaly, can also mask the presence of awareness. In addition, patients in a minimally conscious state may display inconsistent behaviour, making it difficult to interpret their responses, and they may be not aware for protracted intervals, making it difficult to interpret failure to respond.
Secondly, there is a conceptual problem in the logic of establishing "lack of awareness": absence of evidence (of awareness) is taken as evidence of absence (of awareness). Consequently, on the basis of the current clinical standards, patients who are aware but non-responsive cannot be distinguished from non-aware (vegetative) patients. Clinically, this flaw in logic introduces a category of aware but non-responsive patients for whom a diagnosis of vegetative state is technically appropriate (that is, they show no signs of awareness) but incorrect (in fact, they are aware).
Is there a place for brain imaging as a diagnostic tool?
In recent years, techniques such as positron emission tomography, functional magnetic resonance imaging, and electroencephalography have been used to try to assess residual brain function and consciousness in vegetative patients without relying on motor behaviour. Neuroimaging studies in patients in a vegetative state have shown a consistent reduction in brain metabolism of as much as 50%w22 and reduced basal resting state activity.In addition, unexpected levels of residual cognitive function (such as processing of linguistic and self referential stimuli) are present in both minimally conscious patients and patients in a vegetative state In some of these cases, high level functions (such as learning and actively maintaining information through time) are present, as are awareness and the ability to communicate solely by modulation of brain activity.
The Multi-Society Task Force on PVS states, however, that "neurodiagnostic" tests, although recognised as "providing useful information when used in conjunction with clinical evaluation" are believed to be unable, alone, to "either confirm the diagnosis of vegetative state . . . or predict the potential for recovery of awareness."4
Although we agree that functional neuroimaging cannot confirm a diagnosis of vegetative state, it is increasingly clear that functional neuroimaging can be used to rule out a diagnosis of vegetative state and may even yield information about prognosis. Indeed, limited data on prognosis show that quantitative measurements of brain activity—in particular, activations beyond primary sensory cortices—are positively correlated with recovery from the vegetative state.
Conclusion
Disorders of consciousness remain challenging to manage because of our superficial understanding of the phenomenon of consciousness and its neural mechanisms. Two main strategies seem promising for reducing the consistently high misdiagnosis rate. Firstly, behavioural assessments need to be conducted more thoroughly and by trained staff (a neurologist or another healthcare professional who has been trained to use the formalised assessments mentioned previously).Secondly, we believe that the inclusion of recommendations for the use of functional neuroimaging techniques in revised guidelines will increase the detection of covert signs of awareness in the very circumstances susceptible to misdiagnosis. In addition, these techniques can be used to explore the degree of mental life possible after severe brain injury,w24 thus tackling the medically and ethically important question "what is it like to be in a vegetative state?" In a minority of cases, these techniques may even allow the patients to interact with their environment and to some extent let their voice be heard.
• Summary points
• A careful clinical history, physical examination, and referral to evidence based guidelines when ordering imaging will reduce unnecessary studies,Although head computed tomography is less expensive, faster, and more readily available, magnetic resonance imaging is better at assessing most neurological conditions• Head computed tomography is recommended when urgent decisions are needed but has a low yield in transient neurological episodes.When vascular lesions are suspected, as in transient ischaemic attack, computed tomography angiography or magnetic resonance angiography of the head and neck is indicated
• Treat most patients with low back and neck pain only conservatively; reserve imaging for those with red flag features and those who fail conservative treatment and are candidates for surgical intervention Practitioners may need to assess the need for further testing and possible interventions in patients with incidental findings seen on neurological imaging
A guide to imaging for common neurological problems
BMJ 16 August 2010
• Patients with headache, transient neurological episodes, symptoms after minor head trauma, and neck and low back pain often present to general practitioners and emergency room physicians. The examining doctor may be uncertain whether neurological imaging is needed. In this article, we discuss indications for imaging and tests that would be most useful in these scenarios. provides a walk through of representative images from patients with headache or minor head trauma, showing normal and abnormal findings.
When is neuroimaging needed after minor head trauma?
Minor head trauma and post-concussion syndrome are common (see box 1 for defining criteria). In the United States, 128 per 100 000 population present to emergency rooms with minor head trauma each year. Doctors worry about fracture, contusion, and space occupying haemorrhage or enlarging haemorrhage—lesions that might signify the need for neurocritical care or neurosurgical intervention .Large prospective cohort studies show that the probability of neurologically important conditions or problems requiring neurosurgical intervention is less than 10% and 1%, respectively, so not all patients with minor head trauma need imaging, and those who do need to be identified.
• Box 1 DefinitionsMinor head trauma
• Blunt head trauma within the past 24 hours and Glasgow coma scale score of 13 or more, together with resultant loss of consciousness or amnesia or disorientation• Post-concussion syndrome
• Less than four weeks between head trauma with loss of consciousness and development of symptoms plus at least one symptom from three of the following categories:
• Headache, dizziness, fatigue, noise intolerance
• Irritability, depression, anxiety, emotional lability
• Reduced subjective concentration or memory, or intellectual difficulties without neuropsychological evidence of marked impairment
• Insomnia
• Reduced alcohol tolerance
• Preoccupation with above symptoms and fear of brain damage, with hypochondriacal concern and adoption of sick role
IndicationsTwo large prospective studies evaluated patients who underwent head computed tomography after minor head trauma. They each produced a set of clinical decision rules aimed at detecting those patients who need neurosurgical intervention or are harbouring brain injuries (box 2). The presence of any of the findings in either rule set identified all at risk patients (100% sensitivity for each set). Many experts therefore advocate head computed tomography only for patients with positive findings from either set of rules.
• Box 2 Decision rules from New Orleans and Canadian studiesNew Orleans criteria (initial Glasgow coma scale score 15)*
• Headache
• Vomiting
• Age >60 years
• Drug or alcohol intoxication at the time of trauma or evaluation
• Persistent anterograde amnesia
• Trauma above the clavicle
• Seizure
• Canadian head computed tomography rule (initial Glasgow coma scale score 13-15)*
• Glasgow coma scale score <15 two hours after injury
• Suspected open skull fracture
• Signs of basal skull fracture
• Two or more episodes of vomiting
• Age >65 years
• Cannot remember anything that happened for at least 30 minutes before the trauma
• Dangerous mechanism
• The lack of studies on neuroimaging in post-concussion syndrome probably means that clinicians use a similar approach to the one they use with acute minor head trauma. Imaging is avoided if brain injury is not apparent on clinical examination and the diagnosis of post-concussion syndrome is clear.
What modalities are used?Computed tomography of the head is faster than magnetic resonance imaging (5-10 minutes v 20 minutes) and detects more pathology. For example, a retrospective analysis of 100 patients showed that 58% of small traumatic subdural haematomas were detected only by magnetic resonance imaging.6 Although the improved prognostic information and data altered medical management, they did not necessarily change surgical management.6
Advanced imaging techniquesA variety of new imaging modalities can improve the detection of intracranial lesions and help explain physical findings. This may be particularly valuable in trauma patients, in whom imaging results may be normal despite a poor clinical state. Diffusion tensor imaging measures the directionality of water movement along white matter tracts. Diffusion tensor imaging abnormalities may correlate with destruction of white matter pathways and help in prognosis.7 A variety of functional and perfusion imaging modalities may show focal or regional abnormalities even when standard imaging is normal.7
How should patients be evaluated after a transient neurological episode?
When a neurological episode is truly transient, in that no residual symptoms, signs, or deficits remain, history taking is the key to distinguishing high risk patients from low risk ones. Table 2 offers an evidence based approach where data are available and gives answers to important questions that may help establish a diagnosis.The initial investigation of a patient with neurological symptoms that have resolved often focuses on distinguishing between two potential causes of the episode: transient ischaemic attack and epileptic seizure. A meta-analysis found a 15% risk of infarction within three months of a transient ischaemic attack, half of which occurred within 48 hours. A seizure may point to a serious underlying lesion or systemic illness, or it may herald status epilepticus.
Suspected transient ischaemic attackFor patients with suspected transient ischaemic attack, urgent imaging of the head and neck vasculature is needed to determine the subsequent risk of stroke and to guide decision making about interventions such as carotid endarterectomy. Head magnetic resonance imaging and head and neck magnetic resonance angiography, or head computed tomography and head and neck computed tomography angiography.
Magnetic resonance imaging is often preferred because it is more likely to detectpathology. An aggregate of 19 studies (1117 patients) showed that magnetic resonance diffusion weighted imaging detected small areas of infarction in 39% of patients ,even when symptoms and signs had resolved and the patient met clinical criteria for transient ischaemic attack.
Suspected seizureMagnetic resonance imaging can identify pathology that may underlie a seizure. Head computed tomography and magnetic resonance imaging both yield abnormal findings in 10% of patients with seizure, and both techniques are recommended by the American Academy of Neurology and American Epilepsy Society). Magnetic resonance imaging with gadolinium is more sensitive than computed tomography, however, for detecting a wide range of causes including tumour ,haemorrhage, infection, inflammation, and developmental anomalies .
Transient neurological episode of uncertain causeIn many cases, the differential diagnosis remains broad even after a thorough history and examination. Such patients might report a variety of symptoms not related to either transient ischaemic attack or seizure, such as dizziness, vertigo, presyncope, and syncope. In this setting, imaging is not recommended when the most likely diagnoses include migraine, benign forms of vertigo, psychogenic spells, and cardiogenic or neurogenic (vasovagal) syncope.
Head computed tomography alone is unlikely to be useful in such patients, although evidence to guide imaging practice is lacking.
In certain circumstances, especially when history and examination are incomplete, incidental imaging findings may steer the clinician away from the correct diagnosis.
For example, a patient with transient neurological symptoms found to have a chronic cerebral infarction by imaging might be improperly diagnosed with a transient ischaemic attack. This patient may not have had a proper cardiac evaluation, which might have shown that the neurological symptoms were caused by a cardiac arrhythmia.
Is imaging always needed for severe headache?
Large prevalence studies have shown that primary headache disorders, such as migraine, affect more than 10% of the population. A large literature review and a prospective evaluation of patients with non-acute headache found intracranial lesions—such as haemorrhage, raised pressure, infection, or tumour—in less than 1% of patients with a normal neurological examination.Box 3 outlines features of the patient’s history that should alert a clinician to serious pathology. Expert reviews suggest that investigations are needed only if the history indicates a worrisome entity or the neurological examination is abnormal. A systemic review showed that the presence of four of the following features associated with headache was strongly suggestive of migraine and made imaging unnecessary: pulsatile quality, duration four to 72 hours, unilateral location, nausea and vomiting, and headache that is disabling.
• Clinical features of headache that warrant imaging12 13 14
• Acute thunderclap* headache, cluster-type headache, or undefined (not a known primary headache disorder) quality of headache• Accelerating (or new) pattern of headache, including during pregnancy
• Aggravation of headache by exertion or Valsalva-like manoeuvre
• Onset of headache after age 50
• Non-classic visual symptoms or other aura
• Vomiting
• Other focal neurological symptoms or signs (such as aphasia, neglect, or hemiparesis) or an abnormal neurological examination*
• Current or alternative medical systemic illness or signs (such as sinusitis or mastoiditis, HIV, cancer, fever, rash)
• *Particularly predictive of abnormal neuroimaging.1
• The presence of any one symptom must be considered in the context of the entire clinical picture. For example, vomiting associated with other symptoms of migraine should reassure the clinician, whereas vomiting associated with non-migrainous headache is a cause for concern (box 3).
• If the history and examination do not support a diagnosis of primary headache disorder or if red flags are raised, the American Academy of Neurology and the American College of Radiology (http://acsearch.acr.org) recommend imaging to exclude pathology.
The results of a randomised controlled trial suggested that neuroimaging reduces anxiety in patients with headache, which may reduce subsequent costs. This might encourage doctors to request imaging in some patients even if the examination does not suggest pathology.
What imaging technique is most useful?The best test to choose depends on the suspected diagnosis .In patients with acute headache who need rapid diagnosis because of suspected intracranial haemorrhage or cerebral aneurysm, head computed tomography and computed tomography angiography are preferred to evaluate the brain parenchyma and vasculature, respectively.
In most other situations, head magnetic resonance imaging with gadolinium is preferred because it provides better anatomical detail and special sequences to probe various pathologies.The addition of gadolinium increases the sensitivity of disease detection and lesion description, particularlywhen intracranial infection, tumour, or other inflammatory processes are suspected.
Advanced imaging techniquesRecent studies have shown syndrome specific alterations in regional cerebral metabolism and blood flow that may offer further insight into the aetiology of headache.
When is imaging needed in patients with neck and low back pain?
Neck and low back pain is ubiquitous and affects most people at some point in their life. A large population based study found a 31% three month prevalence in adults in the US. In such patients, imaging often shows non-specific or non-diagnostic findings, such as mild to moderate degenerative spine disease (fig 2F-G).A meta-analysis of randomised controlled trials found that lumbosacral spine imaging does not improve outcomes in patients with isolated low back pain without an indication of a serious underlying condition. Although studies evaluating diagnostic imaging for neck pain are lacking, the same conclusion would probably hold true. The American College of Physicians and the American Pain Society recommend conservative treatment for patients with neck pain and back pain that is not associated with neurological signs or concerning features.
Imaging should be reserved for patients with red flag features on history or physical examination. These include previous trauma; constitutional symptoms such as unexplained fever or weight loss; systemic disease or cancer; and motor, sensory, or sphincteric deficits. In the absence of concerning features, imaging is generally deferred for at least six weeks while conservative treatment is instituted, because most patients improve during this time.
Choice of imaging modalityMagnetic resonance imaging optimally evaluates the spinal cord, nerve roots, intervertebral discs, ligaments, bony elements, and soft tissues—features that are not well delineated by computed tomography (fig 2F, 2G, and 2I). Gadolinium enhanced magnetic resonance imaging is indicated when neoplastic, infectious, or inflammatory conditions are suspected (fig 2I).
Computed tomography may be useful when bone integrity needs to be assessed, such as in a patient with a history of trauma, osteomyelitis, or metastases (fig 2H). Although plain radiographs allow evaluation of spinal alignment and stability (with flexion and extension images) and the basic integrity of the bony elements, they have limited use in many settings because of poor anatomical and spatial resolution.
How should incidental findings be interpreted and managed?
Magnetic resonance imaging detected incidental findings in about 14% of brain imaging studies in a large adult Western population. Common incidental findings include asymptomatic infarcts (fig 2J), benign tumours (fig 2D), aneurysms (fig 2K), and small white matter lesions (fig 2L). Practitioners must decide whether to disregard such findings as clinically unimportant, obtain additional images, refer to a specialist, or manage directly. The direct and indirect costs of incidental findings, especially in patients in whom imaging is overused, is unknown.Small white matter lesionsWhite matter disease comprises a diverse set of cerebral pathologies, but it is usually chronic cerebral ischaemic microangiopathy when found incidentally in elderly people and those with risk factors for cerebrovascular disease.29 The incidence of this type of white matter disease increases with age and, when moderate to severe, increases the risk of stroke in the next four years fivefold; it is also associated with a decline in cognitivefunction.30
Asymptomatic infarctsAsymptomatic infarcts increase the risk of subsequent stroke threefold. Their presence should prompt an evaluation of risk factors for cerebrovascular disease, such as hypertension, hyperlipidaemia, and diabetes, and the need for antiplatelet treatment should be considered. Vascular and cardiac studies, such as non-invasive angiography of the head and neck, echocardiography, and ambulatory electrocardiography, may also be considered.
Benign tumoursThe most common incidental benign tumours are meningiomas and pituitary adenomas; if smaller than 1 cm in diameter they do not usually become problematic. Although controversial, small meningiomas (<2 cm) do not generally warrant subsequent imaging unless they lie in potentially high risk locations (such as parasellar or at the cerebellopontine angle). In contrast, one small study found that pituitary adenomas less than 1 cm in diameter carry a 15% risk of enlargement, so they may warrant further evaluation (neuroendocrinological studies and neurological investigations such as formal visual field testing).
AneurysmsA large prospective study reported that small aneurysms in the anterior circulation (anterior cerebral, middle cerebral, and internal carotid derived arteries; <7 mm) have a 0% risk of subsequent rupture at five years. However, in a retrospective review of 152 patients with aneurysmal subarachnoid haemorrhage, the ruptured aneurysms were less than 7 mm in diameter in 100 (65.7%) of these patients. Thus, controversy exists regarding the best management of patients with small aneurysms discovered incidentally, and whether it is good practice to perform serial imaging.
Until consensus emerges it is reasonable to avoid serial imaging in such patients with the caveat that an irregularly shaped aneurysm, hypertension, and young age (<50 years) may increase the risk of subsequent rupture. Patients with any other type of cerebral aneurysm are best referred to a specialty clinic where the patient’s history and aneurysmal factors will guide risk assessment and subsequent investigation and management.
New Chronic Pain Guidelines
The recommendations apply to patients with chronic noncancer, neuropathic, somatic, or visceral pain. The taskforce focused on interventional diagnostic procedures including diagnostic joint block, nerve block, and neuraxial opioid trials.Medscape April 1, 2010
The new guidelines detail
• ablative techniques,• acupuncture,
• blocks,
• botulinum toxin,
• electrical nerve stimulation,
• epidural steroids,
• intrathecal drug therapies,
• minimally invasive spinal procedures,
• pharmacologic management,
• physical therapy,
• psychological treatment, and
• trigger point injections.
The taskforce defines chronic pain as pain of any etiology not directly related to neoplastic involvement associated with a medical condition or extending in duration beyond the expected temporal boundary of tissue injury and normal healing and adversely affecting the function or well-being of the individual.
Medscape April 1, 2010
Drugs for chronic pain include anticonvulsants, antidepressants, benzodiazepines, N-methyl-D-aspartate receptor antagonists, nonsterioidal anti-inflammatories, opioid therapy, skeletal muscle relaxants, and topical agents. The taskforce discusses each in detail and recommends strategies for monitoring and managing adverse effects and patient compliance.
New Diagnostic Criteria for Multiple Sclerosis
RecommendationsClinically definite multiple sclerosis can be confirmed if a MRI is performed at any time demonstrating dissemination in space and showing at least 1 or more asymptomatic gadolinium-enhancing and nonenhancing lesions.
Patients without any enhancing or with all enhancing lesions would require a new MRI to demonstrate new T2 or gadolinium-enhancing lesions.
Medscape February 12, 2010
Patients with an abnormal MRI performed at any time, but not showing dissemination in space or time, would require follow-up imaging.
"We recommend 1 dissemination in space criterion," the authors note. This would represent 1 or more asymptomatic T2 lesions in 2 or more of 4 locations considered characteristic for multiple sclerosis in previous MRI criteria — juxtacortical, periventricular, infratentorial, and spinal cord.
"
We recommend 2 dissemination in time criteria," they add, and call for the presence of at least 1 or more asymptomatic gadolinium-enhancing and nonenhancing lesions irrespective of the time of the scan and the presence of a new T2 or gadolinium-enhancing lesion compared with a previous scan.
Asked by Medscape Neurology to comment on the new criteria, Gary Birnbaum, MD, from the Minneapolis Clinic of Neurology in Golden Valley, Minnesota, raised some concerns about the effect these recommendations could have.
"The proposed criteria are less stringent than other criteria, possibly increasing the sensitivity of detecting conversion to clinically definite multiple sclerosis, but possibly decreasing the specificity of the changes," he said. "This will need to be monitored carefully."
Increasing the Risk for Misdiagnosis
Dr. Birnbaum says the data supporting early treatment of multiple sclerosis are good; however, he suggests that not all patients with clinically isolated syndrome need, or necessarily benefit from, treatment with disease-modifying therapies.This will need to be monitored carefully.
"Indeed, recent long-term data show that more than 30% of persons with 'high-risk' MRI changes do not have significant disease after 20 years of follow-up," he said. "There may not be a great urgency for establishing a diagnosis of clinically definite multiple sclerosis in a significant proportion of individuals with clinically isolated syndrome, especially if the specificity of criteria are less stringent — increasing the risk of misdiagnosis."
Dr. Birnbaum points out there are many disease-modifying therapies already approved for the treatment of patients with clinically isolated syndrome. "If a physician feels there is a need to begin treatment early, conversion to clinically definite multiple sclerosis is not necessary to prescribe such agents," he said.
More Study Needed
Dr. Birnbaum emphasizes that having less stringent MRI criteria for establishing disease progression may not be necessary and could increase the risk of treating people with diseases other than multiple sclerosis.Ben Thrower, MD, a neurologist and senior medical advisor for the Multiple Sclerosis Foundation, said he agrees there are risks involved in early diagnosis. This could lead to inaccurate diagnosis or patients starting therapy with expensive and inconvenient medications, he suggests.
However, Dr. Thrower also acknowledges the benefits of early diagnosis, including peace of mind for patients and families dealing with unexplained symptoms. He points to data suggesting fewer relapses and new MRI lesions in those who start therapy sooner.
"
These new proposed guidelines offer the possibility of using a single MRI to fulfil diagnostic criteria," Dr. Thrower said. "If the brain MRI shows lesions with and without active inflammation, this would demonstrate dissemination in time and space."
Dr. Thrower says the new criteria will need to be studied further and compared with existing McDonald criteria to determine whether they are sufficient.
Dr. Montalban and his team acknowledge that testing these criteria in new, prospectively followed clinically isolated syndrome cohorts is needed.
They also point out that scans in most studies have been performed on 1.5-Tesla or lower field-strength scanners using conventional T2-weighted spin echo or fluid-attenuated inversion recovery sequences.
They note, "The impact of 3 Tesla or higher field MRI findings or other MRI sequences on diagnosis will have to be considered in the future."
Transient loss of consciousness—initial assessment, diagnosis, and specialist referral: summary of NICE guidance
Initial assessment
At any stage, including initial presentation, if the person has sustained an injury or has not made a full recovery of consciousness, or if transient loss of consciousness is secondary to a condition that needs immediate action, use clinical judgment to determine appropriate management and the urgency of treatment.
Ask the person with suspected transient loss of consciousness, and any witnesses (try to contact these by phone if necessary), to describe what happened:
-Before the event (circumstances, posture, prodromal symptoms)
-During the event (appearance and colour; movement; any tongue biting or injury; and duration of transient loss of consciousness)
-During the recovery period (confusion, weakness down one side).
Assess and record:
-Details of any previous transient loss of consciousness-Medical history and any family history of cardiac disease
-Current medication
-Vital signs
-Lying and standing blood pressure if appropriate
-Other cardiovascular and neurological signs.
Important abnormalities in a 12 lead electrocardiogram in patients with transient loss of consciousness
Atrial arrhythmia (sustained)
Inappropriate persistent bradycardia
Conduction abnormality (for example, complete right or left bundle branch block or any degree of heart block)
Left or right ventricular hypertrophy
Long QT interval (corrected >450 ms) and short QT interval (corrected <350 ms)
Pathological Q waves
Ventricular pre-excitation
Any ventricular arrhythmia (including ventricular extrasystoles)
Brugada syndrome
Paced rhythm
Any abnormalities in ST segment or T wave, especially abnormal T wave inversion
Making a judgment based on initial assessment
Red flags: refer the patient within 24 hours for a specialist cardiovascular assessment by the most appropriate local service if the person has any of the following:
-Transient loss of consciousness during exertion
-New or unexplained breathlessness
-Heart failure
-Family history of sudden cardiac death in people younger than 40 years and/or an inherited cardiac condition
-A heart murmur
-Any of the important electrocardiographic
If the patient has no features suggesting an alternative cause of transient loss of consciousness (note that brief seizure activity can occur during uncomplicated faints), diagnose uncomplicated faint or situational syncope, or suspect orthostatic hypotension when there are suggestive features as follows:
-Uncomplicated faint (the “three Ps”): posture (a faint may follow prolonged standing or the patient may have a history of similar episodes prevented by lying down); provoking factors (such as a medical procedure); prodromal symptoms (such as sweating before transient loss of consciousness)
-Situational syncope, in which syncope is clearly and consistently provoked by straining (such as with micturition), coughing, or swallowing
-Orthostatic hypotension, in which the history is typical and lying and standing blood pressure (with repeated measurements while standing for three minutes) confirms orthostatic hypotension.
Suspect epileptic seizures :
-A bitten tongue-Head turning to one side during transient loss of consciousness
-No memory of abnormal behaviour even though such behaviour has been witnessed by someone else before, during, or after transient loss of consciousness
-Unusual posturing
-Prolonged jerking of limbs
-Confusion after transient loss of consciousness.
-Prodromal déjà vu, or jamais vu (a feeling that something is happening for the first time, despite knowing rationally that it has happened before)
Specialist cardiovascular assessment and diagnosis
Conduct a specialist cardiovascular assessment and thereby assign a suspected cause of syncope (such as structural heart disease, cardiac arrhythmia, carotid sinus syncope, or neurally mediated syncope) or unexplained syncope. Offer further testing as directed in the recommendations or other tests as clinically appropriate.
Offer exercise testing (unless contraindicated) when transient loss of consciousness has been experienced during (not after) exercise.
Do not offer a tilt test to people who have a diagnosis of vasovagal syncope on initial assessment, because tilt testing has a low predictive accuracy in this population.
For people with suspected cardiac arrhythmia as a cause of syncope, or with unexplained syncope (including those aged 60 years and older who have had a negative result for carotid sinus massage), offer ambulatory electrocardiography (and do not offer a tilt test before this procedure).
Choose the type of ambulatory electrocardiography according to the frequency of the patient’s transient loss of consciousness:
• -Several episodes a week: offer Holter monitoring (up to 48 hours), then if no further loss of consciousness occurs offer an external event recorder
• - Episodes every one to two weeks: offer an external event recorder, then if necessary offer an implantable event recorder
• - An episode less than once every two weeks: offer an implantable event recorder.
Further information on the guidance
The causes of transient loss of consciousness vary, and people experiencing it may come under the care of a range of healthcare professionals, from general practitioners and ambulance staff to emergency department clinicians and specialists in cardiology or neurology. The lack of a clear pathway may contribute to misdiagnosis and inappropriate treatment. The guideline tackles these problems and provides recommendations for initial assessment, specialist referral, and further tests, so that a correct diagnosis can be reached quickly, efficiently, and cost effectively..
Determining the mechanism for transient loss of consciousness in an individual requires collection of evidence (from a detailed history, clinical assessment, and appropriate investigations) and interpretation of each piece of evidence in overall context with clinical reasoning. The guideline shows how this can best be achieved in the most cost effective way and gives indicators for diagnosis of some causes of transient loss of consciousness; however, it does not seek to replace clinical judgment or make recommendations on treatment.
What’s new
The NICE guideline emphases the importance of obtaining an account of the event from the person who had transient loss of consciousness and from any witnesses and of using this information to make initial diagnoses.Central to the guideline is the requirement that all people with transient loss of consciousness should have 12 lead electrocardiography, preferably using an automated machine.
In line with the epilepsy guideline, we recommend against the routine use of electroencephalography in the investigation of transient loss of consciousness.
Further investigations focus particularly on the use of ambulatory electrocardiography devices, with the type of device selected being dependent on the frequency of previous episodes of transient loss of consciousness, thereby moving away from the often indiscriminate use of Holter monitors
If Your Stomach Is Bigger, Is Your Brain Smaller?
Visceral fat is associated with smaller brain volume.large study shows a strong inverse association between visceral obesity and low total brain volume. The authors hypothesize that inflammation resulting from adipose tissue or adipose tissue–derived hormones (e.g., leptin, ghrelin) is the possible mechanism for this relationship, which is stronger in visceral adipose tissue.
Low brain volumes have also been shown to be related to insulin resistance, but this relation does not explain the current finding. This study's findings are another reason to be concerned about obesity and about whether some psychopharmacologic medications that cause weight gain have adverse cerebral consequences.
Journal Watch Psychiatry July 19, 2010
• Oral and Transdermal Estrogen Therapy and Risk for Stroke
• Low-dose transdermal delivery was not associated with elevated risk.• In the Women's Health Initiative randomized trials, postmenopausal oral estrogen therapy raised risk for stroke. However, whether this risk is elevated among transdermal estrogen users is unknown. In this population-based nested case-control study, investigators used a large U.K.

database to assess risk for stroke associated with use of oral and transdermal hormone therapy among 870,000 women (age range, 50–79) during the 20-year study period.
Researchers identified 16,000 women who received diagnoses of first stroke and matched them with 60,000 randomly selected controls.
After adjustment for multiple confounders, current use of any dose of oral estrogen or oral estrogen–progestogen was associated with elevated risk for stroke, compared with nonuse. In contrast, current use of low-dose transdermal estrogen ( 50 µg) was not associated with elevated stroke risk, but higher-dose transdermal estrogen was associated with elevated stroke risk (rate ratio, 1.89).
Comment: These results suggest that low-dose transdermal estrogen therapy — unlike oral estrogen — is not associated with stroke. The findings are also biologically plausible. As the authors note, "The transdermal route avoids the first pass effect in the liver and therefore reduces the induction of hepatic protein synthesis of clotting factors and various inflammation markers."
The results also are consistent with those of prior studies in which oral estrogens, but not transdermal estrogens, raised risk for venous thromboembolism (JW Gen Med Jul 1 2008). However, keep in mind that this is an observational study with the potential for unidentified confounders and is not a randomized trial; indeed, that elevated risk for stroke was associated with oral estrogen use was not established until the randomized Women's Health Initiative was performed.
Journal Watch General Medicine June 22, 2010
Nothing to Cough About: A Treatment for Involuntary Laughing and Crying
Dextromethorphan with quinidine appears safe and effective for a syndrome seen in neurological disorders.Some patients with neurological disorders such as multiple sclerosis (MS) and amyotropic lateral sclerosis (ALS) experience involuntary episodes of laughing, crying, or both. These episodes have been variously termed as pathologic laughing and crying, emotional incontinence, and pseudobulbar affect.
Controlled studies have demonstrated the efficacy of 30 mg of the cough medicine dextromethorphan (Dm), combined with 30 mg of the antiarrhythmic quinidine (Q; an inhibitor of cytochrome P450 2D6) to increase Dm levels. These researchers ascertained the safety and effectiveness of a much lower Q dose (10 mg). In a randomized, controlled, manufacturer-funded study, 326 patients with MS or ALS received 30 or 20 mg Dm combined with 10 mg Q (DmQ 30/10 or DmQ 20/10) or placebo for 12 weeks.
Episode frequency diminished in all groups, but significantly more so with either DmQ dose than with placebo (47%–49% reductions compared with placebo). The 12-week mean change in daily episode rate was –3.9 to –4.1 for DmQ and –3.0 for placebo. Remission (no episodes in the final 14 study days) was seen in significantly more patients receiving DmQ than placebo (about 50% vs. about 30%). Social functioning and mental health improved with the higher Dm dose. No significant electrocardiographic changes were seen.
Comment: This underrecognized clinical syndrome might affect patients with many neurological disorders. Although some publications have reported efficacy of selective serotonin reuptake inhibitors for this condition, more data exist on this new combination, approved in October 2010 by the FDA at the 20/10 dose. DmQ was well tolerated, with dizziness, nausea, diarrhea, and urinary tract infections reported as more frequent with the higher (unapproved) DmQ dose than with placebo. The medication appears both safe and effective, but clinicians might still use SSRIs initially because of their familiarity. Could DmQ be used for other indications, such as irritability or depression? These potential therapeutic uses could be explored.
Journal Watch Psychiatry November 15, 2010
Failure of adequate trials of two (or more) tolerated,appropriately chosen, and appropriately used antiepileptic drug regimens (whether administered as monotherapies or in combination) to achieve freedom from seizures.
Drug-Resistant Epilepsy
nejm.org 926 september 8, 2011
Although drug resistance may “remit” over time (at a rate of 4% per year among adults and a higher rate
among children), seizure relapse is common, suggesting a fluctuating course.
Other consistent clinical predictors of drug resistance include a high number or frequency
of seizures in the early phase of the disorder and the presence of a known,often structural cause of the epilepsy, particularly hippocampal sclerosis.
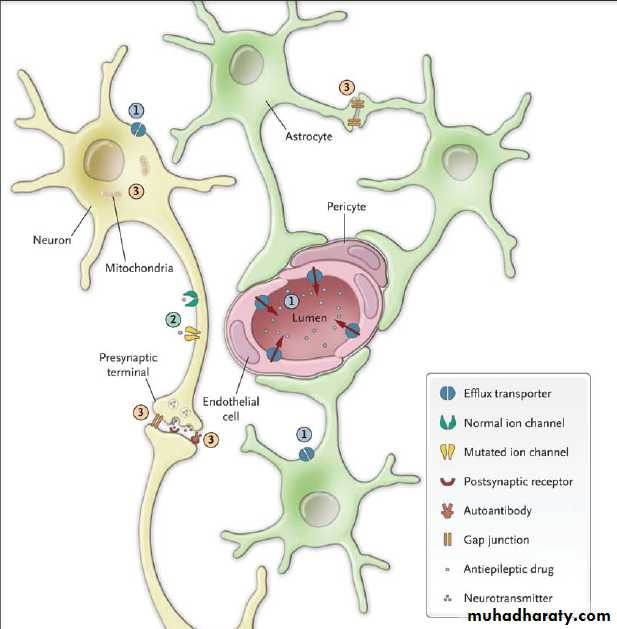
Hypothesized Biologic Mechanisms of Drug Resistance in Epilepsy.
Hypothesized Biologic Mechanisms of Drug Resistance in Epilepsy.The diagram illustrates the molecular locations at which the mechanisms are hypothesized to operate. At locations Labeled 1 is overexpression of efflux transporters in capillary endothelial cells that constitute the blood–brain barrier.At the location labeled 2 is altered expression or function of neuronal voltage-gated ion channels that are known Targets of antiepileptic drugs. At locations labeled 3 are mechanisms not targeted by current antiepileptic drugs,such as electrical coupling through gap junctions, mitochondrial dysfunction, and autoantibodies to neurotransmitter
receptors.
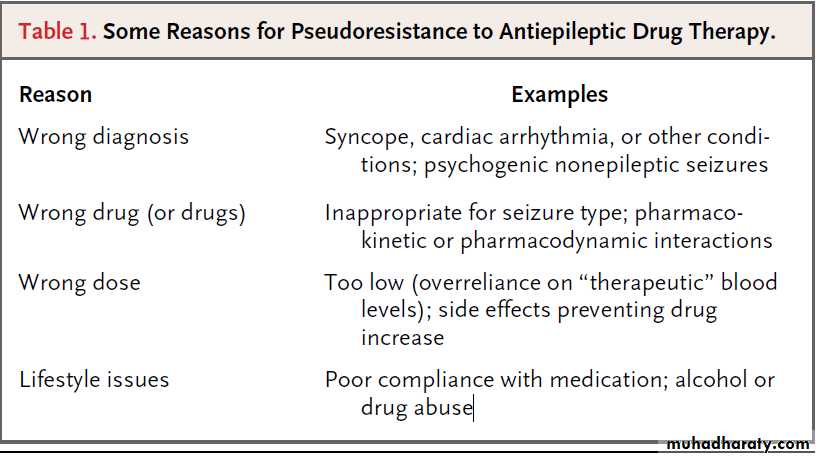
Principles of Management
Ruling out PseudoresistancePseudoresistance, in which seizures persist because
the underlying disorder has not been adequately
or appropriately treated, must be ruled out or corrected before drug treatment can be considered
to have failed. This phenomenon may arise in a
number of situations (Table 1), of which misdiagnosis
of epilepsy is probably the most common.
Conditions that frequently mimic epileptic seizures include vasovagal syncope, cardiac arrhythmias,metabolic disturbances, and other neurologic disorders with episodic manifestations (e.g., transient ischemic attacks and migraine). Psychogenic, nonepileptic seizures are estimated to account for more than 25% of adult cases of apparently drug-resistant epilepsy.
Failure of drug therapy may also result from an
inadequate understanding of the pharmacologicproperties of antiepileptic drugs, particularly their
range of clinical efficacy and pharmacokinetic
characteristics. These properties are summarized
in Table 2 in the Supplementary Appendix. Because
the spectrums of activity vary among antiepileptic
drugs, incorrect classification of the syndrome
or seizure type can lead to treatment failure
or even seizure aggravation.
Table 3 in the Supplementary Appendix shows the latest recommendations for seizure classification. Phenytoin, carbamazepine, gabapentin, oxcarbazepine, vigabatrin, tiagabine, and pregabalin can worsen absence epilepsy and myoclonic seizures. Lamotrigine can also exacerbate some myoclonic epilepsy syndromes.
Another problem is that an antiepileptic drug may fail to control seizures satisfactorily because it is not prescribed at the optimal dosage. This may result from an injudicious reliance on monitoring of serum drug concentrations;
a “therapeutic range” can be interpreted as
dictating dosage adjustment without adequate
clinical correlation.
Other possible causes of pseudoresistance may
be related to the patient’s lifestyle or behavior,particularly insufficient adherence to the therapeutic
regimen, which may even contribute to increased
risks of illness and death.36 Abuse of alcohol
and recreational drugs can cause seizures.
Sleep deprivation and stress are also common seizure-precipitating factors.
General Approach
Once a patient’s epilepsy is recognized to be drug
resistant, a personalized treatment plan should be
formulated to limit any cognitive deterioration or
psychosocial dysfunction. It is a good idea to mention
early in the course of treatment that total
freedom from seizures may not be attainable. This
approach may help pave the way for a later palliative
strategy if that proves to be necessary.
Non drug therapy such as epilepsy surgery should be considered. Patients should also be informed of the risk of sudden, unexpected death in epilepsy;appropriate precautions might include nocturnal supervision, although hard evidence is lacking that this is likely to be lifesaving. Conditions commonly associated with treatment-resistant epilepsy,such as anxiety, depression, and cognitive and memory disturbances, should be recognized and treated.
Combination Therapy
Generating robust clinical evidence of suitablecombinations of antiepileptic drugs has been challenging because of the large number of possible
combinations of drugs and dose ranges. The strongest evidence in favor of synergism comes from
nonrandomized, controlled studies involving adults
who received a combination of sodium valproate
and lamotrigine for partial-onset and generalized
seizures (class III evidence); these observations
are supported by animal models.
Other combinations that are sometimes recommended, largely on the basis of anecdotal reports or studies with small samples, include valproate with ethosuximide for absence seizures (class IV evidence) and lamotrigine with topiramate for a range of seizure types (class IV evidence).
One strategy for combination therapy that has
been advocated is a pharmacomechanistic approachbased on the drugs’ differing modes of
action, although high-quality data to support this
strategy are lacking. Data from studies in animals
suggest that administering two drugs that act on
the same pharmacologic pathway, such as sodiumchannel
blockade, is less effective than administering
two drugs with different mechanisms of action.
The possibility that combining two drugs that act by blocking neuronal voltage-dependent sodium channels may not be clinically useful was
first reported in 1975 by Cereghino and colleagues,
who treated 47 cognitively impaired patients
with phenytoin, carbamazepine, and phenobarbital
sequentially and found that combinations
with phenobarbital were more effective than phenytoin combined with carbamazepine (class III).
Deckers and colleagues undertook acomprehensive review of the available data from studies in animals and humans and concluded that combinations involving a sodium-channel blocker and a drug with GABAergic properties appeared to be particularly beneficial. However, the successful licensing of the latest sodiumchannel blockers, lacosamide and eslicarbazepine, as adjunctive treatment was based on data from studies in which the majority of patients were
already taking drugs with similar mechanisms of
action, such as carbamazepine or lamotrigine.
Combinations of drugs that act primarily by blocking
voltage-dependent sodium channels (e.g., lamotrigine and carbamazepine) may be more likely
to be associated with neurotoxic effects, such as
dizziness, diplopia, and ataxia (class III evidence).
Latest Developments in Drug Therapy
In double-blind, randomized trials, the efficacyof adjunctive treatment with modern antiepileptic
drugs has been disappointingly small,46 fueling
continuing efforts to develop new compounds.
Although a reduction in seizure frequency of 50%
or more is generally accepted as demonstrating
efficacy for regulatory purposes, the clinical relevance
of such an improvement to the overall
health status of patients is limited47 and freedom
from seizures should remain the goal of treatment.
In the past 2 years, two new sodium-channel
blockers, lacosamide48 (in the United States and Europe).and eslicarbazepine (in Europe), have been
licensed for use in adults with partial seizures with
or without secondary generalization (class I evidence).
Rufinamide has shown effectiveness as a
treatment for the Lennox–Gastaut syndrome in infants
and children (class I evidence).
Vigabatrin was recently licensed in the United States as an adjunctive treatment for complex partial seizures in adults and as monotherapy for infantile spasms in children from 1 month to 2 years of age (class I evidence), although it has been available elsewhere for many years. Stiripentol has been approved under the orphan-drug procedure in Europe for the treatment of Dravet’s syndrome, a rare childhood epilepsy syndrome (class I evidence).
The U.S. and European regulatory authorities
have recently granted approval to retigabine (ezogabine in the United States and Canada) as anadjunctive treatment for refractory partial seizures
with or without secondary generalization
in adults (class I evidence). Unlike other antiepileptic drugs, this drug acts by opening potassium channels.
Other drugs that are undergoing phase 3 trials include brivaracetam (which, like Levetiracetam,binds to the synaptic vesicle protein 2A molecule) and perampanel, which modulates glutamate neurotransmission mediated by α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA).
Nondrug Therapy
Patients who meet the criteria for having drugresistantepilepsy should be evaluated early for
surgical treatment, particularly if they have a surgically
remedial syndrome, such as unilateral hippocampal
sclerosis or other resectable lesions.
The decision to offer surgical treatment requires
an individualized risk–benefit assessment that includes
consideration of the pros and cons of additional
trials of antiepileptic drugs.
A range of surgical procedures can be performed, depending on the indication. The prototype is anterior temporal lobectomy, which has been shown in a randomized,controlled trial56 to be superior to continued medication in providing long-term relief
from seizures in up to 70% of adults with drugresistant
temporal-lobe epilepsy (class I evidence).
Other potentially curative procedures include resection
of structural lesions (lesionectomy) such
as glial tumors and vascular malformations (class III).
Even when magnetic resonance imaging reveals no lesions in patients with temporal or extratemporal epilepsy, resection may be supported by findings from functional imaging (ictal single-photon-emission computed tomography or interictal positron-emission tomography) with or without invasive electroencephalographic monitoring, although the outcomes of surgical treatment in such cases tend to be less favorable than those in lesional cases (class III).
Palliative procedures, which are intended to
disrupt the pathways important for the propagation of epileptiform discharges and thus reduce the frequency and severity of seizures, may be considered when resection of the seizure-generating region is not possible. Corpus callosotomy is usually performed in children with clinically significantlearning disabilities and severe generalized
epilepsy, particularly when the disorder
causes atonic seizures that are associated with
frequent falls and subsequent injuries; adults may
also benefit but to a lesser degree (class III evidence).
Multiple subpial transection is a less commonly performed procedure that is reserved
for situations in which the epileptogenic focuscannot be removed because of close proximity to
eloquent cortex. This procedure is usually performed in children in conjunction with corticalresection, which makes it difficult to assess its specific efficacy (class IV evidence). Hemispherectomy or functional hemispherotomy, performed in both children and adults, is a more dramatic procedure in which an extensively diseased and epileptogenic cerebral hemisphere is removed or functionally disconnected (class IV evidence).
The vagus-nerve stimulator is a multiprogrammable
pulse generator that is implanted in the
patient’s upper chest and delivers electrical current
to the vagus nerve, usually the left nerve, in
the neck.61 The device has been approved for use
as an adjunctive therapy for adults and adolescents
older than 12 years of age whose partial-onset
seizures are resistant to antiepileptic medication,
although the response is modest (class I evidence).
The ketogenic diet (a high-fat, low-protein,
low-carbohydrate diet) is used in children withdrug-resistant epilepsy. A randomized, controlled
trial showed that the number of seizures fell by
more than 50% in approximately half of children
after 1 year on the diet (class II evidence).62 The
diet seems to be effective for all seizure types.
The major problem is adherence to the restrictive
(and unpleasant) dietary regimen. Therefore,
a modified Atkins diet is under evaluation as a
potential alternative in adults and for environments
in which strict supervision is unavailable
A range of new approaches to the treatment of
drug-resistant epilepsy are under active investigation.
Those that are in advanced clinical development
include technology-based approaches that
use intracranial and extracranial treatment systems,
which typically provide either electrotherapy
or pharmacotherapy and which in some cases may
be automatically administered when a seizure is
detected by sensors.
New and Emerging Ther apies
One such intracranial device,which delivers scheduled electrical stimulation
bilaterally to the anterior nucleus of the thalamus,was studied in a multicenter, double-blind,randomized trial involving 110 adults with drugresistant focal epilepsy (class I evidence).The group receiving electrical stimulation had a 29%
greater reduction in seizures than the control group(no stimulation). After 2 years of open-label use in
a continuation phase of the trial, the median reduction in seizure frequency was 56%; 54% of patients had a seizure reduction of at least 50%, and
14 patients were seizure-free for at least 6 months.
The Food and Drug Administration Neurological
Devices Panel recently recommended approval of
the device.
Another device undergoing a phase 3 clinical trial in adults involves a “closed-loop system”:
when the device detects epileptiform activity,
it also delivers electrical stimulation to the
site of this activity. The limited published clinical
data regarding the device appear to be favorable.66
Other promising therapeutic strategies in more
preliminary stages of development include stereotactic radiosurgery, stem-cell therapy, and gene
therapy.
Complementary and Alternative Therapies
Complementary and alternative medicine encompassesdiverse medical and health care systems,
practices, and products that are not generally considered part of conventional medicine as practiced,
for example, in the United States. Such therapies
are widely used (up to 50% of people with epilepsy
in developed countries may have used them at some
time), although patients may be reluctant to spontaneously report their use to physicians.
The majority of patients use complementary or alternative therapies not for seizure control but for
general health purposes or for symptoms that
could be indicative of coexisting conditions, such
as depression, or of treatment-related adverse
events. Clinicians should therefore specifically
inquire about the use of such therapies and the
underlying reasons, which may prompt additional
evaluations and treatments.
To date, no complementary or alternative therapy
has been shown to be effective for epilepsy in
multicenter, double-blind, controlled trials. Contrary
to the popular belief that “natural is safe,”
such therapies can be harmful to people with epilepsy, and natural products pose the greatest
risk of side effects, interactions with antiepileptic
drugs, and seizure exacerbation.
Table 4 in the Supplementary Appendix lists the characteristics and potential benefits and risks of natural products that are reportedly used by people with epilepsy. For example, ginkgo biloba, often taken to enhance cognitive function, has been suspected of lowering serum concentrations of phenytoin and valproate by inducing the hepatic drug-metabolizing cytochrome P-450 enzyme CYP2C1971, and has anecdotally been reported to exacerbate seizures.
St. John’s wort, commonly taken to relieve symptoms of depression, may also, at least in theory, interact with antiepileptic drugs through induction of cytochrome P-450 enzymes, although data in support of a clinically relevant interaction are lacking. Nonetheless, some natural products and their constituent compounds have been shown to have mechanisms of action that are relevant to epilepsy or to have anticonvulsant properties in animal models and are undergoing further preclinical evaluation.
nejm.org 926 september 8, 2011


