
Gynaecology
Dr. Raghad
Lec 24 - Intersex Disorders
DR. RAGHAD - LEC 3


1
Intersex disorders
Embryology:
There are three factors which determine an individual’s sexual development. These
are:
1. The effect of the sex chromosomes on the differentiation of the gonad.
2. The proper functioning of the differentiated testis.
3. The response of the end organ to this testicular function.
The testes carry out their intrauterine function by producing two substances:
1.
Testosterone: stimulates the development of the Wolffian duct, which
differentiates into the internal male genitalia and also to the masculinization of
the cloaca. The manner in which testosterone, produced by developing testes, is
utilized to bring about masculinization of the cloaca is through conversion of
testosterone to dihydrotestosterone through the action of the enzyme 5α-
reductase. (Wolffian structures are capable of utilizing testosterone directly and
are therefore independent of 5α-reductase activity).
2.
Müllerian inhibiting substance (MIS): inhibits the development of the Müllerian
structures which are always present and capable of development. MIS may have
unilateral action so that each testis appears to produce the hormone, which
results in regression of the Müllerian structures on its own side.
Sexual development may be abnormal in the following circumstances:
1. Sex chromosome abnormalities interfering with testicular differentiation (like
46X/46XY mosaicism) giving rise to one of the forms of gonadal dysgenesis.
2. Anatomical testicular failure (incapable to produce testosterone) or
enzymatic testicular failure (biosynthetic defect of testosterone).
3. The end organs may be incapable of utilizing testosterone because of 5α-
reductase deficiency or because the androgen receptor is abnormal and
therefore testosterone cannot bind to the cell wall (androgen insensitivity).
4. The production of Müllerian inhibitor may be deficient, leading to the growth
of Müllerian structures in an otherwise normal male.

2
5. In a genetic female masculinization of the external genitalia may result in
cases of excessive androgen production in utero, for example, congenital
adrenal hyperplasia.
6. Rarely, in a genetic female, genes capable of producing the H-Y antigen may
be found on an autosome, leading to the condition known as the 46XX male.
7. True hermaphroditism, i.e. the presence of testicular and ovarian tissue in
the same individual, may be present and such patients are commonly
genetically female with mosaicism, though genetic male variants also exist.
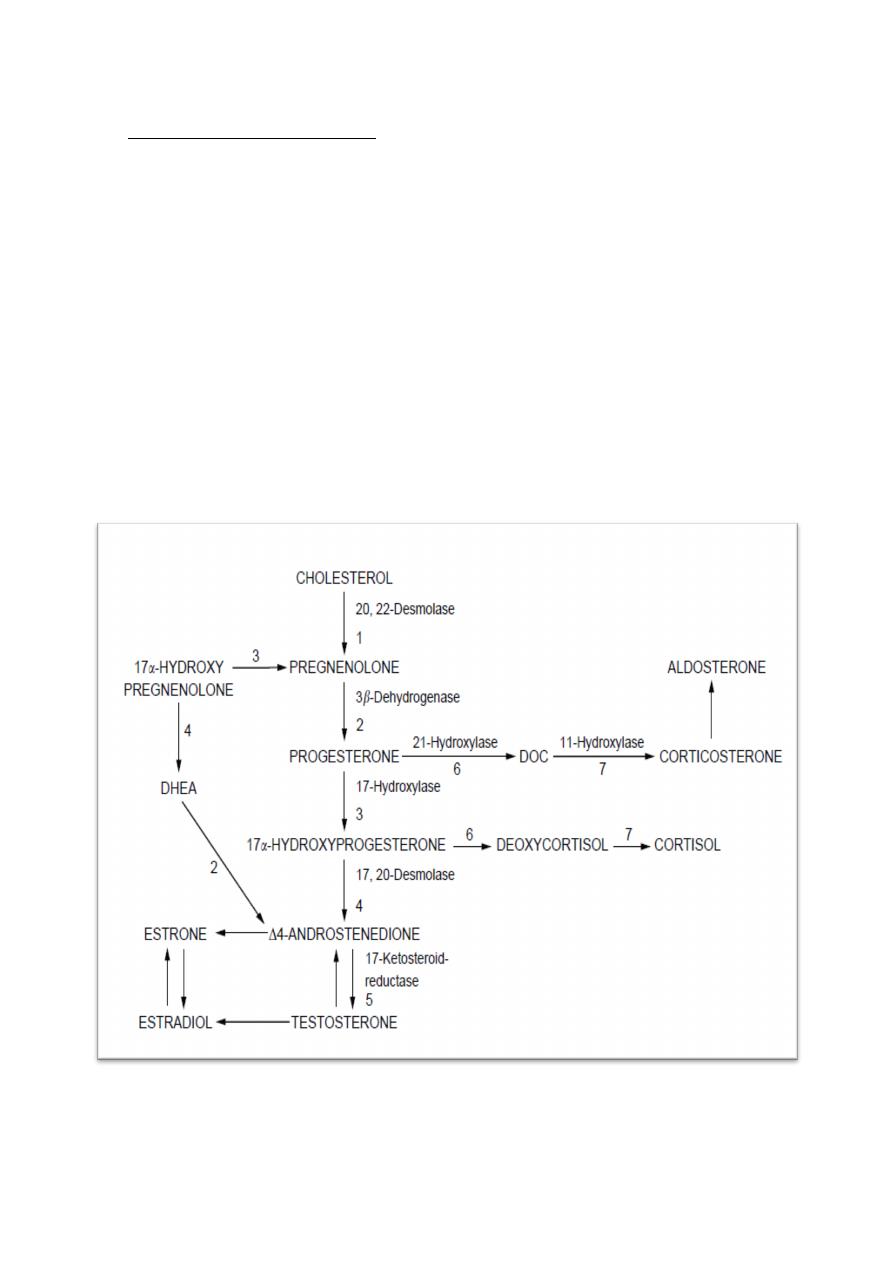
Congenital Adrenal Hyperplasia:
CAH is the most common cause of female intersex and is an autosomal
recessive disorder resulting in enzyme deficiency related to the biosynthesis of
cortisol and aldosterone.
The commonest enzyme defect is 21-hydroxylase deficiency (incidence is
between 1 in 5000 - 1 in 15,000) which results in failure of conversion of 17α-
hydroxyprogesterone to desoxycortisol and progesterone to desoxycorticosterone.
In 21-hydroxylase deficiency, which accounts for 90% of cases of CAH, the deficiency
results in an increase in progesterone and 17α-hydroxyprogesterone, which is
therefore converted to androstenedione and subsequently to testosterone.
Presentation: Affected females are born with enlargement of the clitoris and
excessive fusion of the genital folds (ambiguous genitalia), which obscure the
vagina. Thickening and rugosity of the labia majora are evident, they bear some
resemblance to the scrotum. The uterus, fallopian tubes and vagina are always
present. These clinical changes of masculinization are secondary to the elevated
levels of androgens as a result of the enzyme defect.
In some infants a dangerous salt-losing syndrome may arise because of
associated aldosterone deficiency and the child may die of wasting and vomiting
within a few weeks of life if the diagnosis is not appreciated.

3
Management of ambiguous genitalia in new born infants:
ü
Counseling the parents that the child is healthy, but there is a developmental
anomaly of the genitalia.
ü
Initial examination of the child, if fail to identify a palpable gonads it is most
likely that the child is female and the likelihood of CAH may be raised.
ü
Investigation of a suspected case of CAH should include:
a) Karyotyping, which may be performed on cord blood and results are
rapidly obtained (within 24 hrs).
b) Measurement of 17α-hydroxyprogesterone in blood, which will be
elevated in 21-hydroxylase deficiency.
c) Examination of electrolytes to check the possibility of a salt-losing
syndrome, in such cases, sodium and chloride may be low and
potassium is raised.
d) Pelvic ultrasound to reveal the presence of a uterus and vagina. This is
not only reassuring to the parents, but highly indicative of the correct
diagnosis.
ü
The immediate management of such a child should always be undertaken by
or in cooperation with the paediatrician. Cortisol or one of its related
synthetic compounds must be given to suppress adrenocorticotrophic
hormone secretion. If the child is a salt loser then salt loss must be very
carefully controlled.
ü
Surgical clitoral reduction may be undertaken, and simple labial fusion can be
treated simply by surgical division.
Note:
other causes of ambiguous genitalia in genetically female infants
include: androgen secreting tumors that have occurred in pregnancy (which
result in verilization of the fetus especially luteomy), krukenburg tumor and
polycystic ovaries, or intake of exogenous progestogens which is rare.
ü
CAH if not treated at birth (seen at puberty), then management depend on
the patients' sex of rearing. Those who have reared as females are unlikely to
have major masculanization of the external genitalia, thus cosmetic surgery is
required and cortisol is given which will be sufficient to stimulate
development of secondary sexual charecteristics and withdrawal bleeding.
While those who are reared as male with gross masculanization of external
genitalia and good functioning phallus might be allowed to continue in this
gender role with total hysterectomy and oopherectomy and subsequent
testosterone replacement.

4
XY Females - End Organ Insensitivity:
1. 5α-Reductase deficiency:
Normal masculinization of the external genitalia requires the
conversion of testosterone to dihydrotestosterone by 5α-reductase. Although
the Wolffian structures respond directly to testosterone, in the presence of
5α-reductase deficiency a male infant will have poor masculinization of
external genitalia. The patient has the following characteristic:
• 5α-Reductase deficiency is a familial disorder due to an autosomal
recessive gene, so the evidence of other similar affected members in
the family often assists the diagnosis.
• Uterus, tubes and upper vagina will always be absent since MIS
production will be normal.
• There is mild to moderate degree of genital masculinization, so most
children are initially placed in the female role.
• At puberty, however, the testes produce increased amounts of
testosterone and there is greater virilization, perhaps to an extent that
the patient may wish to change the gender role from female to male.
• The female gender role will often be a better one for such patients.
2. Androgen insensitivity
This condition occurs when there is partial or complete absence of
androgen receptors. The patient has the following charecteristics:
• Karyotype is 46 XY.
• Presented after puberty as primary amenorrhea despite normal breast
development.
• Normal vulva, with absent or scanty pubic and axillary hair.
• Absent uterus with short blind vagina.
• Testes are present in the lower abdomen, in the inguinal canal or
occasionally in the labia.
• Endocrine investigations reveal normal male range of testosterone,
while estrogen level is generally within the range where normal male
and normal female values overlap.
• About 5% of patients will develop gonadal cancer, which is sufficiently
high to warrant gonadectomy.
• Hormone replacement therapy is required following gonadectomy.
5
True Hermaphrodites:
o
It is rare.
o
Karyotype in 58% 46XX, and in 13% 46XX/XY.
o
Distribution of the gonads is interesting, the commonest combination is
for an ovotestis to be present on one side and an ovary on the other. but
the presence of testis on one side and an ovary on the other is not
uncommon being almost as frequent as the previous combination.
Ovotestis may also be bilateral or combined with a testis.
o
Diagnosis of true hermaphroditism can only be made by GONADAL
BIOPSY to demonstrate that ovarian and testicular tissue are both
present.
o
Sex of rearing is determined on the functional capability of the external
genitalia, after which, inappropriate organs are removed.
Diagram of the enzyme steps necessary to convert cholesterol through its various intermediate
stages to aldosterone, cortisol and testosterone