
Dr. Mushtak Gherbawe
Lec. 2
METABOLIC
DISORDERS
Wed
17 / 12 / 2014
Published by : Ali Kareem
2014 – 2015
مكتب اشور لالستنساخ
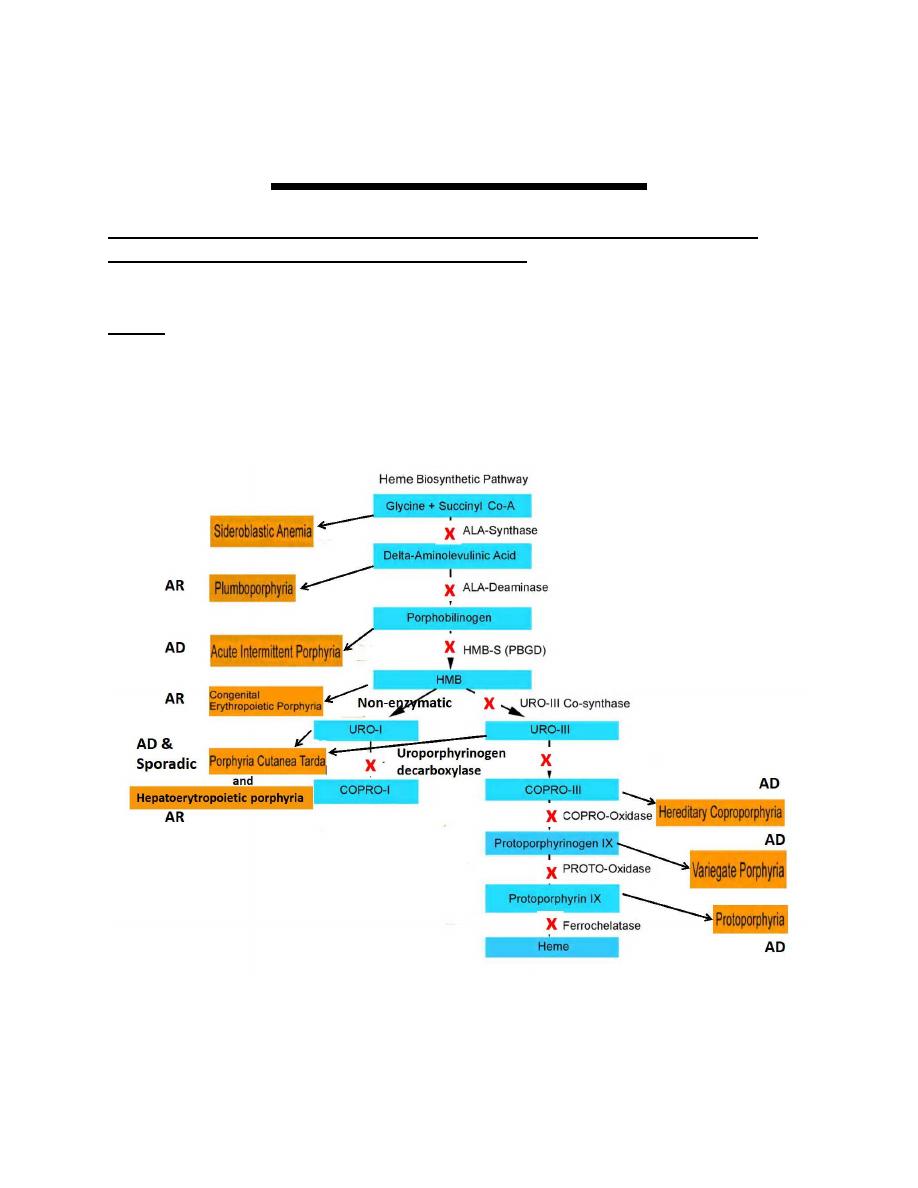
Porphyrias
The porphyrias are group of metabolic disorders, resulting from the
deficiency of a specific enzyme in the heme biosynthetic pathway
leading to overproduction of intermediates to an abnormally high level
leading to a disease.
Heme It is the prosthetic group ( nonprotein component of a conjugated
protein) of: Hemoglobin, Myoglobin, and the cytochromes.

They are commonly categorized as either
� Hepatic or Erythropoietic, depending on the site of excess porphyrin
Production
� Acute or Cutaneous : according to their clinical manifestations.
1.Hepatic
They are six, three of the six present with acute porphyrias and they are
1-Acute intermittent porphyria (AIP),
2-Hereditary coproporphyria (HCP),
3-ALA-dehydratase porphyria (ADP)
present during adult life.
� The fourth hepatic is, (4)-porphyria cutanea tarda (PCT), presents with
cutaneous photosensitivity, and blistering skin lesions.
� The fifth and sixth (5)-hereditary coproporphyria and (6)-variegate
porphyria can present with either symptom pattern or with a mixed
picture.
2.Erythropoietic porphyrias
Congenital erythropoietic porphyria (CEP) and erythropoietic
protoporphyria (EPP), present with cutaneous photosensitivity, and
usually at birth or in early childhood.

The Acute Porphyrias (relapsing and remitting)
� Present with acute attacks of neurologic manifestations (neurovisceral)
in the
form of:
� Abdominal , mental, and neurologic symptoms.
� They are typically triggered by drugs and other exogenous factors,
Present during adult life
The Cutaneous Porphyrias (Photosensitive skin manifestations)
� Due to excess production and accumulation of porphyrins in the skin.
� These porphyrins are activated by long-wave ultraviolet light (UVA) with
generation of oxygen radicals that damage the skin. Leading to:
� Pain, erythema, bullae, erosions, hirsutism and hyperpigmentation ,
that occur predominantly on areas of the skin that are exposed to
sunlight.
� They usually present at birth or in early childhood.
Porphyria Cutanea Tarda (PCT)
Hepatic Porphyria, it is either sporadic or AD inheritance with cutaneous
photosensitivity manifestation.
It is the most common porphyria due to deficiency of uroporphyrinogen
decarboxylase (UROD), and can be either:
Sporadic (80%) (type 1) or
Familial (20%) (type 2)
CLINICAL FEATURES
Blistering skin lesions that appear most commonly on the backs of the
hands. May also occur on the forearms, face, legs, and feet (sun exposed
areas). These rupture and crust over, leaving areas of atrophy and scarring
lesions. Skin friability and small white papules termed milia are common,
Hypertrichosis and hyperpigmentation, especially of the face, are especially

troublesome in women. Occasionally, the skin over sun-exposed areas
becomes severely thickened, with scarring and calcification that resembles
systemic sclerosis.
Neurologic features are absent.
Causes
Enzyme defects mentioned or risk factors that can both cause and
exacerbate the disease as alcohol, excess iron, HFE gene defect(
hemochromatosis)i.e. iron overload disorder (common to occur in PCT) and
Hepatitis C virus …etc (all decrease hepcidin production by hepatocytes),
leading to increase iron absorption and deposition in the liver, causing
oxidative damage to liver cells, resulting in oxidized uroporphyrinogen that
inhibit the activity of hepatic UROD. It is also increase the uptake of iron in
liver cells, leading to further oxidation of uroporphyrinogen by therelease of
activated oxygen radicals.
Diagnosis
Uroporphyrin and heptacarboxyl porphyrin in urine is diagnostic.
Increased isocoproporphyrin in feces confirm diagnosis.
Treatment
Patients are advised to discontinue alcohol, estrogens, iron supplements,
or other contributing factors.
A course of phlebotomies is the preferred treatment. Because iron stores
in PCT are not markedly increased, removal of only 5 to 8 units of blood at
1- to 2-week intervals is usually sufficient.
A course of low-dose chloroquine or hydroxychloroquine is usually effective
when repeated phlebotomies are contraindicated.

Acute intermittent Porphyria
It is a hepatic porphyria with autosomal dominant inheritance.
Common in Scandinavia and Great Britain (North Europe).
2
nd
most common porphyria.
Pathophysiology
The disorder remains asymptomatic in the great majority prior to
puberty. It present in conditions that increase need of heme synthesis
in the liver and the specific enzyme cannot meet the demand.
Common precipitating factors include:
Endogenous and exogenous steroids, certain drugs, alcohol ingestion,
Low-calorie diets, usually instituted for weight loss,Infections, Surgery.
The fact that AIP is almost always latent before puberty suggests that :
Adult levels of steroid hormones are important for clinical expression.
Symptoms are more common in women, suggesting a role for estrogens or
progestin.
Acute porphyrias are sometimes exacerbated by exogenous steroids,
including oral contraceptive preparations containing progestin
Increased carbohydrate intake can ameliorate attacks.
Clinical Manifestations
Abdominal pain, the most common symptom cramping. Ileus, abdominal
distention, nausea, vomiting, constipation are common.
Abdominal tenderness, fever, and leukocytosis are usually absent
because the symptoms are neurologic rather than inflammatory.
Sensory loss; dysuria; and urinary retention are characteristic.

Tachycardia, hypertension, restlessness, tremors, and excess sweating are
due to sympathetic overactivity.
Mental symptoms such as anxiety, insomnia, depression, hallucinations,
and paranoia can occur.
Seizures can be due to neurologic effects or to hyponatraemia. Treatment
of seizures is difficult because most antiseizure drugs can exacerbate AIP
(clonazepam may be safe).
Hyponatremia results from hypothalamic involvement and SI ADH or
from electrolyte depletion due to vomiting, diarrhea, poor intake, or
excess renal sodium loss.
When an attack resolves, abdominal pain may disappear within hours, and
paresis begins to improve within days and may continue to improve over
several years.
DIAGNOSIS
ALA and PBG levels are substantially increased in plasma and urine,
especially during acute attacks, and become normal only after prolonged
latency.
TREATMENT:
During acute attacks, narcotic analgesics may be required for abdominal
pain, and phenothiazines are useful for nausea, vomiting, anxiety, and
restlessness. benzodiazepines are probably safe in low doses if a minor
tranquilizer is required.
Carbohydrate loading
Usually with intravenous 10% dextrose in water daily), may be effective
in milder acute attacks of porphyria (without paresis, hyponatremia, etc.).
Hemin(Hematin)
Is more effective and should be used initially for severe attacks and for
mild attacks that do not respond to carbohydrate loading within 1
–2
days.
It is the hydroxide of heme, leading to decrease heme biosynthesis
through
–ve feedback and prevent further accumulation of heme
precursors.

Hemochromatosis
Hereditary Hemochromatosis
Hemochromatosis, HFE gene-related (type 1) Hemochromatosis,
Non-HFE gene-related:
• Juvenile H (hemojuvelin mutations) (type 2A) AR inheritance.{AR}
• Juvenile H (hepcidin mutation) (type 2B) {AR}
• Mutated transferrin receptor 2 TFR2 (type3) {AR}
Mutated ferroportin 1 gene, SLC11A3 (type 4) AD inheritance.
Acquired Iron Overload (2ndry)
Iron-loading anemias Thalassemia major Sideroblastic anemia Chronic
hemolytic anemias
Chronic liver disease Hepatitis C Alcoholic cirrhosis especially when
advanced, Nonalcoholic steatohepatitis.
Others Transfusional and parenteral iron overload ,dietary iron
overload,porphyria
cutanea tarda ….etc.
Hereditary Hemochromatosis
Normally the HFE gene with the other genes or receptor mentioned
especially hepcidin peptide, prevents excessive iron absorption and
storage in normal people (control iron absorption), so any mutation or
abnormality lead to excess iron absorption.
The excess
iron is stored in the body’s tissues and organs, particularly in
the liver, heart, pancreas, endocrine glands, skin, and the joints.

Prevalence
HFE-associated hemochromatosis mutations are among the most
common inherited disease
It is most common in northern Europeans 1 in 10 persons are heterozygous
carriers and 0.3
— 0.5% are homozygotes.
Expression of the disease is effected by,
•Alcohol consumption
•Dietary iron intake,
•Blood loss associated with menstruation and pregnancy,
•Blood donation......etc.
So because of that only 30% of homozygous men and only 1% of the
women develop iron overload
— related disease.
Symptoms usually appear between ages 40 and 60.
The non-HFE-associated forms of hemochromatosis are rare.
Pathophysiology
Normal total body iron content is about 2.5 g in women and 3.5 g in men.
Absorption of iron is approximately 1 mg/day in men and 1.5 mg/day in
menstruating women (Premenopause).
In hemochromatosis, absorption is greater than body requirements and
amounts to 4mg/day or more.
Hemochromatosis may not be recognized until total body iron content is
more than 10g, or often several times greater.
Symptoms and Signs
Iron overload, leads to fibrosis and organ failure. However, organ
damage is slow.
Fatigue and nonspecific systemic symptoms often occur early.
The major clinical manifestations.
1- Cirrhosis of the liver 95%
20 to 30% of patients with cirrhosis develops hepatocellular carcinoma.
Liver disease is the most common cause of death.

2• Hyperpigmentation 70% Due to deposition of melanin and iron in the
dermis. It is usually generalized, but may be more pronounced on the face,
neck, extensor aspects of the lower forearms, dorsa of the hands, lower
legs, and genital regions, as well as in scars.
3• Diabetes mellitus, (bronze diabetes) 65%
4• Arthritis, 45%
Chondrocalcinosis The joints of the hands, especially the second and
third metacarpophalangeal joints, are usually the first joints involved, a
feature that helps to distinguish the chondrocalcinosis associated with
hemochromatosis from the idiopathic form.
progressive polyarthritis involving wrists, hips, ankles, and knees also
occur.
Acute brief attacks of synovitis may also occur.
5
.
Hypogonadotropic hypogonadism 45%
Occurs in both sexes and, include loss of libido, impotence, amenorrhea,
testicular atrophy, gynecomastia, and sparse body hair.
These changes are primarily the result of decreased production of
gonadotropins due to impairment of hypothalamic-pituitary function by
iron deposition.
6
• Cardiomyopathy and Heart Failure, 15%
The most common manifestation is congestive heart failure.
The heart is diffusely enlarged.
Cardiac arrhythmias and varying degrees of atrioventricular block.
Cardiomyopathy with heart failure is the 2nd most common cause of
death.
7
•Adrenal insufficiency, hypothyroidism, and hypoparathyroidism are
rare manifestations.

Diagnosis
The association of (1) hepatomegaly, (2) skin pigmentation, (3) diabetes
mellitus, (4) heart disease, (5) arthritis, and (6) hypogonadism OR SOME
OF THEM should suggest the diagnosis.
� Serum ferritin Elevated levels (>200 ng/mL in women or > 300 ng/mL in
men) But not diagnostic because it is also elevated in inflammatory liver
disorders (e.g., chronic viral hepatitis, nonalcoholic fatty liver, alcoholic liver
disease), cancer, certain systemic inflammatory disorders (e.g., RA), or
obesity.
� Serum iron (usually > 300 mg/dL).
� % transferrin saturation; levels usually > 50%).
� Total iron-binding capacity ( μg/dL):Decrease.
� Gene assay widespread availability.
� Liver biopsy iron concentration and hepatic iron index is measured.
Also establishes or excludes the presence of hepatic cirrhosis, which is the
critical factor for prognosis.
� High-intensity MRI
Increased density of the liver due to iron deposition can be demonstrated
by CT or MRI, and, with improved technology,
MRI has become more accurate in determining hepatic iron concentration.
It is a noninvasive alternative for Diagnosis.
Treatment:
The therapy of hemochromatosis involves
1- Removal of the excess body iron
2- Supportive treatment of damaged organs.
•lron removal is best accomplished by weekly or twice-weekly phlebotomy
of 500 mL.
•Each 500 mL (1unit of blood) contains 200—250 mg iron, and up to 25 g
iron or more may have to be removed.
•weekly phlebotomy may be required for 1—2 years, and it should be
continued until the serum ferritin level is under 50 g/L.

•Thereafter, phlebotomies are performed at appropriate intervals to
maintain ferritin levels between 50 and 100 g/L. Usually one
phlebotomy every three months.
•Chelating agents such as deferoxamine, when given parenterally,
remove 10
—20 mg iron per day, which is much less than that mobilized by
once-weekly phlebotomy.
•Phlebotomy is also less expensive and safer for most patients.
•However, chelating agents are indicated when anemia or
hypoproteinemia is severe enough to preclude phlebotomy.
•Oral iron chelating agent, deferasirox (Exjade), has recently become
available.
•Alcohol consumption should be severely eliminated because it increases
the risk of cirrhosis in hereditary hemochromatosis nearly tenfold.
•The management of hepatic failure, cardiac failure, and diabetes mellitus
is similar to conventional therapy for these conditions.
• Loss of libido and change in secondary sex characteristics are managed
with testosterone replacement or gonadotropin therapy
Prognosis
Life expectancy is improved by removal of the excessive stores of iron and
maintenance of these stores at near-normal levels. The 5-year survival rate
with therapy increases from 33 to 89%.
Screening
All adult first-degree relatives of patients with hemochromatosis should be
tested.
The life expectancy of those effected and treated before the development
of cirrhosis is normal

WILSON’S DISEASE
It is an autosomal recessive disorder with a frequency of about 1 in
30,000
–40,000, Caused by mutations in the ATP7B gene, a copper-
transporting ATPase Leading to.
� Diminished biliary excretion of copper
� Impaired incorporation of copper into ceruloplasmin. (carries about
70% of the total copper in human plasma) Consequently, copper
accumulates in hepatocytes and extrahepatic sites, particularly in the
central nervous system.
Other organs that are involved:
Kidneys, endocrine organs, heart, and musculoskeletal system.
Clinical manifestations
Are due to copper toxicity leading to
� liver disease
� neurologic abnormalities
� psychiatric abnormalities
Other manifestations
� Renal disease,
� Osteoporosis, osteoarthritis,chondrocalcinosis,
� Cardiac disease,
� Coombs-negative hemolytic anemia.
� Endocrine abnormalities such as infertility, amenorrhea, and
hypoparathyroidism.
� Kayser-Fleischer rings (copper deposits in the outer rim of the cornea)
may be seen.
liver disease
� Involvement vary from simple steatosis to chronic active hepatitis,
fibrosis, fulminant hepatitis, and cirrhosis with portal hypertension.

� Chronic hepatitis, the most common manifestation of hepatic Wilson's
disease, may be difficult to differentiate from chronic hepatitis of other
causes , so Wilson's disease should be included in the differential
diagnosis of any patient with chronic liver disease.
� Liver cancer is uncommon in patients with Wilson's disease.
Motor disorders are typically observed and may include tremors,
dysarthria,
micrographia, pseudobulbar palsy, dysphagia …..etc.
Psychiatric symptoms may range from anxiety to personality disorders to
frank psychosis.
Diagnosis
1- Clinical suspicion, usually because of the presence of hemolytic
anemia, neurologic or neuropsychiatric disease in addition to the liver
disease.
1- Serum ceruloplasmin level, less than 20 mg/dL, raises suspicion
and notdiagnostic
for Wilson’s disease. because:
• May occur in Hypoproteinemia.
•Conversely, ceruloplasmin is an acute phase reactant that may be falsely
elevatedinto the normal range in patients with Wilson’s disease.
The low Serum ceruloplasmin level is due to failure of copper incorporation
intoceruloplasmin, so results in loss of its enzymatic activity and rapid
degradation by plasma proteases and not due to decrease production
2- The free serum copper level is elevated (>25 g/dL).
3- Kayser-Fleischer ring.
4- 24-h urinary copper
excretion usually exceeds 100 μg/24 hr in
symptomatic patients.
5- The gold standard for diagnosis remains liver biopsy with quantitative
copper assays.

Liver biopsy to measure the hepatic copper concentration (>200 tg/g dry
weight of liver).
Liver biopsy and measurement of total urinary copper excretion over a
24-hour period are considered confirmatory tests for the diagnosis of
Wilson’s disease.
Treatment
1- Copper chelating agents, which bind copper and allow it to be
excreted in urine.
2-
a- Penicillamine (chelating agent) was the drug of choice. Dose 1.5
g/day (range 1-4 g). It should always be accompanied by 25 mg/d of
pyridoxine.
Toxic effects of penicillamine occur in one-third of patients and include
rashes, protein-losing nephropathy, lupus-like syndrome and bone marrow
depression, it often worsens existing neurologic disease
b-Trientine (chelating agent) has largely replaced d-penicillamine at a
dose of 1.0 to 1.5 g/day as the initial copper chelator of choice because it is
less toxic.
2- Zinc acetate (Inhibit the absorption of copper and not chelating
agent) at a dose of 50 mg three times daily is an alternative treatment
option for patients:
•Early in the course of the disease, before the development of significant
end-organ damage,
• Maintenance therapy after negative copper balance has been achieved
and storage sites have been depleted
•Preferred in pregnancy.
Treatment must continue for life, even through pregnancy.
Liver transplantation is indicated for fulminant liver failure or for advanced
cirrhosis with liver failure.