Sunday 30 / 11 / 2014
©Ali Kareem 2014-2015
Name
:
______________________________
Class
:
_______________________________
مكتب اشور لالستنساخ
ANTI-ARRHYTHMICS DRUGS
Lecture 6
Total lectures NO. 21
Dr. Samir Matloub

Antiarrhythmic Drugs
Cardiac arrhythmia is abnormality in the rate, regularity or site of origin of the
cardiac impulse, or a disturbance in conduction of the impulse such that the
normal sequence of activation of the atria & ventricles is altered.
Arrhythmia may result from:
1. Disturbances in impulse formation.
2. Disturbances in impulse conduction.
3. Both (combination).
The above 3 points may be caused by many factors: hypoxia, ischemia,
electrolyte imbalance, acidosis, alkalosis, catecholamine exposure, drug
toxicity or presence of scarred or diseased heart tissue.
CARDIAC ELECTROPHYSIOLOGY
The heart contains specialized tissues that exhibit automaticity (can generate
A.P. in the absence of external stimulation). These pacemaker cells differ
from other myocardial cells in showing a slow spontaneous depolarization
during diastole (phase4) caused by the inward current carried by sodium &
calcium.
The depolarization is fastest in the SA node (the normal pacemaker) beating at
a frequency of 60/100 beat/min then the impulse spreads rapidly to the atria
& enters the AV node (which is normally the only conductive pathway
between the atria & ventricles), conduction through the AV node is slow
(0.15 sec) then the impulse propagates over the His-Purkinje system &
invades all parts of the ventricles. Ventricular activation is complete in less
than 0.1 second.
The SA node is the normal pacemaker because it has the steepest or the fastest
rate of phase 4 depolarization. However, the other specialized cells & fibres
of the conducting system are latent pacemakers which may take over if their
automaticity is enhanced.
Most antiarrhythmic drugs have more effects on latent pacemakers than on SA
node pacemaker.
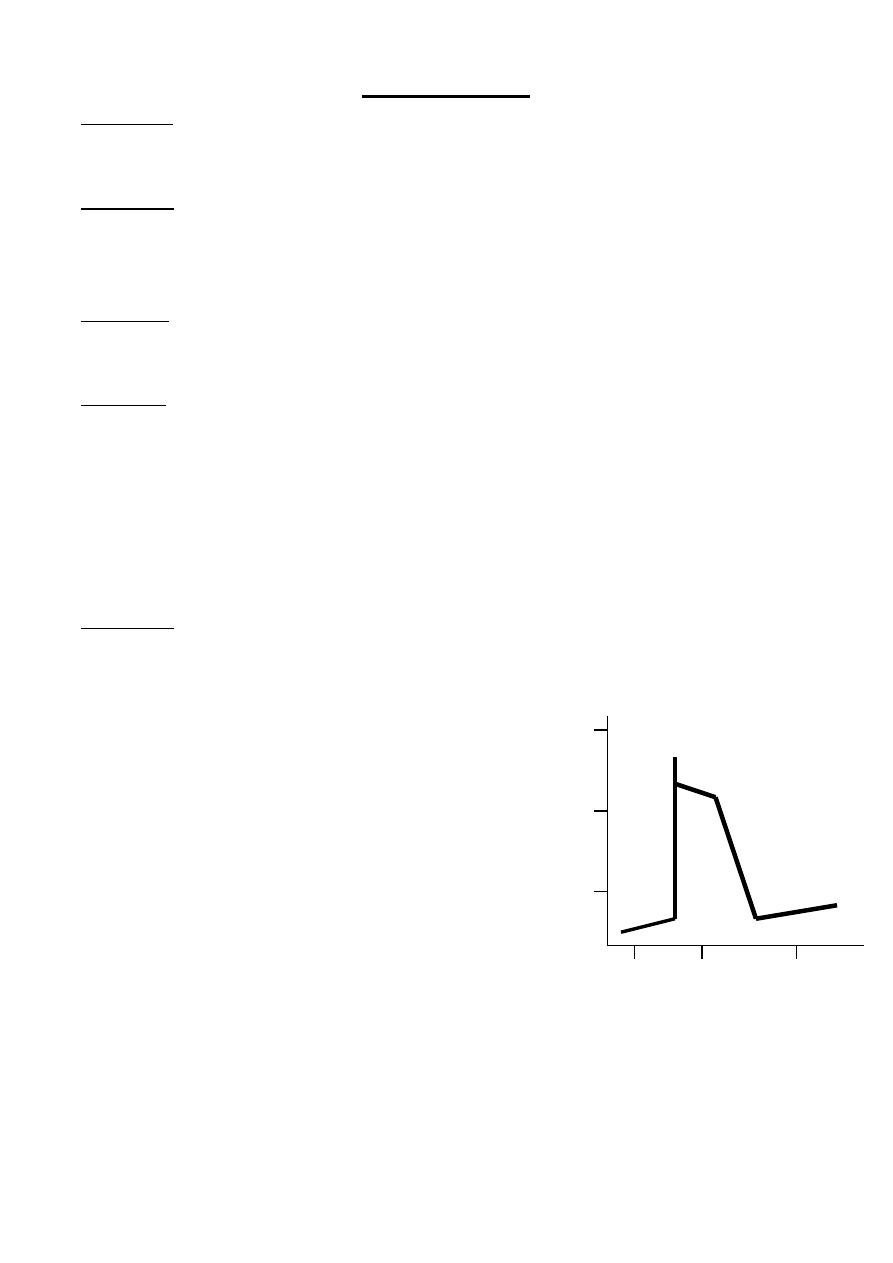
PHASES OF AP
Phase 0: depends on Na
+
current, Na
+
fast channels open, the upstroke
velocity or the max rate of depolarization of phase 0 is conducted V. max the
upstroke ends as the Na
+
channels are rapidly inactivated.
Phase 1: partial repolarization, this rapid initial phase of repolarization is due
to:
1. Inactivation of Na
+
channels.
2. K
+
channels rapidly open & close causing a transient outward current.
Phase 2: plateau; Voltage sensitive Ca
+2
channels open in the same way as
sodium channels but more slowly. The resulting slow inward (depolarization)
current balances the slow (polarizing) outward leak of K
+
.
Phase 3: repolarization (final)
Ca
+2
channels close.
K
+
channels open resulting in an outward current leading to
membrane repolarization.
At the end of repolarization Na
+
& Ca
+2
conc. are high
intracellularly while K
+
is low, this imbalance is corrected by
Na
+
/K
+
ATPase & Ca
+2
is expelled outside by the Na
+
-Ca
+2
exchanger.
Phase 4: forward current occurring in diastole increasing depolarization
results from gradual increase in sodium permeability & also from calcium
inward current (diastolic currents). The spontaneous depolarization
automatically brings the cell to the threshold of the next AP.
+50
Potential (mv) 1
2
0
0
-50 3
4
0 0.5 1
Time (seconds)
A key factor in the pathophys. of arrhythmia & the action of antiarrhythmic
drugs is the relationship between the resting potential of the cell & the AP
that can be evoked in it. This is determined by the availability of sodium
channels ready to allow a rapid sodium influx (activated or open channels).
Fewer Na
+
channels become available as the resting potential becomes less
negative.
Important consequences of reduction in peak Na
+
permeability include:
1. Reduced upstroke volume V. max.
2. Reduced AP amplitude.
3. Reduced excitability.
4. Reduced cond. Velocity.
5. Prolongation of the recovery time which is reflected by an increase in the
effective refractory period.
DISTURBANCES IN IMPULSE FORMATION
The interval between depolarization of a pacemaker cell is the sum of the
duration of AP & the duration of the diastolic interval. Shortening of either
duration leads to an increase in pacemaker rate.
The diastolic interval (most important) is determined by 3 factors:
1. Maximum diastolic potential
2. Slope of phase 4 depolarization.
3. The threshold potential.
Slowing of the rate occurs if:
1. There's more negative max potential (ex.: from -80 to -100 mv).
2. Reduction in the slope of diastolic depolarization.
3. More positive threshold pot (ex.: -65 to -45 mv).
2↓ 3↑
1↓
Vagal stimulation decreases the rate by no.1 & no.2 mechanism, beta blockers
also reduces phase 4 slope

On the other hand ↑ phase 4 depolarization slope leads to ↑ pacemaker rate
which can be caused by hypokalemia, acidosis, B. stimulation & currents of
injury.
Latent pacemakers (ex.: Purkinji fibres) are particularly prone for acceleration
by the above mechanism, however, all cardiac cells may show repetitive
pacemaker activity under appropriate conditions especially if hypokalemia is
also present.
EFFECTS OF DRUGS ON AUTOMATICITY
1. Antiarrhythmic ↓ automaticiy by ↓ slope of phase 4.
2. By raising the threshold to less negative voltage, an effect more prominent
on ectopic pacemaker activities.
DISTURBANCES IN IMPULSE CONDUCTION
In areas of injured myocardium (ex.: ischemia), conduction maybe slow or
refractoriness shortened or both. Resulting in the reentry of aberrant impulse
leading to cardiac arrhythmia. Reentry is the most common cause of
arrhythmia (in which one impulse reenters & excites areas of the heart more
than once). It can occur at any level of the conduction system & may manifest
as one or few extra beats or a sustained tachycardia.
In order for reentry to occur:
1. There must be an obstacle (anatomic pr physiologic) to conduction.
2. There must be a uni-directional block at some point in the circuit.
3. Conduction time around the circuit must be long enough so that the
retrograde impulse doesn't enter ref. Ts. As it travels around the obstacle
(i.e. the conduction time must exceed the eff. Refractory period).
EFFECTS OF DRUGS ON CONDUCTION ABNORMALITIES
Antiarhythmics prevent reentry by
slowing conduction &/or increasing
refractory period to convert a uni-
directional block to a bidirectional
block (by blocking the Na
+
or Ca
+2
current).
CLASSIFICATION OF ANTIARRHYTHMIC DRUGS
They're classified into 4 distinct classes on the basis of their dominant
mechanism of action.

Class I: is subdivided into 3 subgroups according to their effect on the
duration of the AP.
IA drugs which lengthen the duration of the AP.
IB drugs which shorten the duration of the AP.
IC drugs have no effect on duration of AP.
Class II: are the sympathoplegic drugs (reduce adrenergic activity of the
heart) i.e. β-receptors blockers.
Class III: are antiarrhythmic that prolong the AP duration by prolonging
phase 3 repolarization. (K
+
channel blockers).
Class IV: drugs which block the cardiac Ca
+2
currents (i.e. Ca
+2
channel
blockers).
Miscellaneous antiarrhythmics include: digitalis, adenosine, magnesium.
CLASS I
They act by blocking sodium fast channels, these drugs generally cause
slowing of V max, ↓ excitability & ↓ conduction velocity. At therapeutic
doses they have little effect on the resting fully polarized membrane because
they bind more rapidly to open or inactivate channels than to the channels
that are fully repolarized following recovery from previous depolarization
cycle.
Therefore these drugs show greater degree of blockade in tissues that are
frequently depolarizing (ex.: during tachycardia), when the sodium channels
are open. This property called Use dependence or State dependence, which
enables these drugs to block all cells that are discharging at an abnormal high
frequency without interfering with the normal low frequency beating of the
heart.
Class IA: they slow the rate of rise of AP, thus slowing conduction &
↑duration of AP & ↑ the ventricular ERP (effective refractory period). They
have intermediate speed of association with the activated/inactivated sodium
channels & an intermediate rate of dissociation from resting channel.
Example on class IA drugs: quinidine, procainamide & disopyramide.
Class IB: they shorten AP, have little effect on Vmax. They rather ↓ duration
of AP by shortening repolarization (phase3) &, they rapidly interact with Na
+
channels (bind & unbind). Example on this class: Lidocaine, mexiletine,
tocainide & phenytoin.
Class IC: markedly depress Vmax. They cause marked conduction slowing
but have little effect on duration of AP or the ventricular ERP. They bind
slowly to Na
+
. Example on this class: Flecainide, moricizine & propafenone.

Since group IB drugs don't depress the heart in normal dose, they can be used
in heart failure.
The disadvantage of drugs that prolong the QT interval is that they're
proarrhythrogenic.
Group IA drugs in addition to their direct effect occur at higher concentration,
they also express indirect antimuscarinic effects at lower concentration which
antagonize the direct effect. This is shown as tachycardia if quinidine for
example is given to a normal person. This indirect effect is ++ for quinidine,
+ for procainamide & +++ for disopyramide.
Group
Cond.
velocity
ERP
Automaticity
Contractility
Site
of
action
ECG
changes
IA
↓moderate
↑
↓ in all
groups,
effect more
marked on
abn
&
much less
on
SA
node
except
if
diseased or
in overdose
↓
SA &
VA
↑QT
IB
minimal
effect
→
→only in
overdose
VA
→
IC
↓very
marked
↑
↓
SVA
&
VA
→

Quinidine: is an alk. Isolated from cinchona bark. It is the D-isomer of
quinine. It is well absorbed orally (sulphate or gluconate), maximum effect 1-
2 hrs after administration, can be given I.M. (gluconate), 80% bound to
plasma protein, metabolism in the liver & excreted by the kidney.
Pharmacological effects:
1. It blocks sodium channels, ↓ V. max, ↓ conduction velocity & ↑ ERP.
2. Automaticity is ↓ by depression of phase 4 slope, ECG: prolongation of the
QRS & delayed repolariaztion expressed as ↑ QT interval. PR interval may
also be prolonged (little effect). Extra cardiac action: ↓ vascular smooth
mm tone partly by α-receptors blockade→ ↓ peripheral resistance.
Therapeutic uses:
1. SVA (Supra Ventricular Arrhythmia): prevent recurrence of paroxysmal
SVT due to AV nodal or WPW syndrome.
2. Convert atrial flutter or AF to normal sinus rhythm & prevent recurrence of
flutter & AF. Quinidine should not be used without prior digitalization
because of its vagolytic action.
3. Ventricular arrhythmia, PVC to prevent VT after cardioconversion of the
arrhythmia.
Adverse effect:
1. Cardiac toxicity which include:
a. AV block, SA block & even asystole.
b. Exacerbation of the arrhythmia.
c. Myocardial depression: A 50% increase in the QRS complex
duration means you need to decrease the dose promptly.
d. Quinidine syncope: caused by VT of polymorphic form (Torsade
depointes) occurs in patients whose QT interval is increased by the
drug.
e. Quinidine ↑ level of digoxin & can lead to digoxin toxicity.
2. GIT: diarrhea, nausea, vomiting & anorexia are the most common side
effects.
3. CNS-Cinchonism-mild symptoms include: tinnitus, hearing loss, vomiting
& diarrhea. Severe symptoms include: headache, diplopia, altered
conception of colors, confusion & psychosis. These CNS symptoms also
occur with salicylates & quinine.
Contraindications:
Previous allergy, heart failure, hypotension, AV block & sick sinus
syndrome.
Poisoning with digitalis.
Myasthenia gravis, it causes muscular weakness because it blocks sodium
entry to the muscle.

Procainamide:
It differs from the L.A. procain in that it contains an amide structure rather
than an ester linkage that protects it from enzyme hydrolysis.
Pharmacologic effects: are similar to quinidine.
Extracardial effects: when given I.V. it may cause a drop in B.P. due to
peripheral vasodilation.
Therapeutic uses: same as quinidine.
Pharmacokinetics: can be given orally, I.V., I.M.
Peak effect in 1 hr. after oral administration, t
1/2
3hrs.
Elimination by hepatic metabolism and renal excretion.
Renal failure may produce toxicity.
There is marked variation in the rate of acetylation and excretion.
Adverse Effects:
1. Acute procainamide toxicity can produce ventricular arrhythmia, VF and
cardiac depression.
2. Nausea, vomiting, diarrhea and anorexia are common but less than
quinidine.
3. Mental confusion has been reported but less than procain.
4. Hypersensitivity reaction are much more common than quinidine, it
includes:
a. Fever, joint and m pain, and skin rash.
b. Fatal agranulocytosis.
c. A syndrome resembling SLE which is reversible especially in slow
acetylators of the drug.
5. Hypotension when given I.V.
6. Can ppt. glaucoma and urinary retention.
.