
Neoplasia
Cancer is the second leading cause of death in the United States; only cardiovascular diseases exact a higher
toll. Even more agonizing than the mortality rate is the emotional and physical suffering inflicted by
neoplasms. Patients and the public often ask, “When will there be a cure for cancer?” The answer to this
simple question is difficult, because cancer is not one disease but many disorders that share a profound
growth dysregulation. Some cancers, such as Hodgkin lymphoma, are curable, whereas others, such as
pancreatic adenocarcinoma, have a high mortality. The only hope for controlling cancer lies in learning
more about its cause and pathogenesis, and great strides have been made in understanding its molecular
basis. Indeed, some good news has emerged: cancer mortality for both men and women in the United States
declined during the last decade of the twentieth century and has continued its downward course in the 21st.
[1]
The discussion that follows deals with both benign and malignant tumors, focusing on the basic morphologic
and biologic properties of tumors and the molecular basis of carcinogenesis. We also discuss the interactions
of the tumor with the host and the host response to tumors.
Nomenclature
Neoplasia means “new growth,” and a new growth is called a neoplasm. Tumor originally applied to the
swelling caused by inflammation, but the non-neoplastic usage of tumor has almost vanished; thus, the term
is now equated with neoplasm. Oncology (Greek oncos = tumor) is the study of tumors or neoplasms.
Although all physicians know what they mean when they use the term neoplasm, it has been surprisingly
difficult to develop an accurate definition. The eminent British oncologist Willis
[2]
has come closest: “A
neoplasm is an abnormal mass of tissue, the growth of which exceeds and is uncoordinated with that of the
normal tissues and persists in the same excessive manner after cessation of the stimuli which evoked the
change.” We know that the persistence of tumors, even after the inciting stimulus is gone, results from
genetic alterations that are passed down to the progeny of the tumor cells. These genetic changes allow
excessive and unregulated proliferation that becomes autonomous (independent of physiologic growth
stimuli), although tumors generally remain dependent on the host for their nutrition and blood supply. As we
shall discuss later, the entire population of neoplastic cells within an individual tumor arises from a single
cell that has incurred genetic change, and hence tumors are said to be clonal.
A tumor is said to be benign when its microscopic and gross characteristics are considered relatively
innocent, implying that it will remain localized, it cannot spread to other sites, and it is generally amenable
to local surgical removal; the patient generally survives. It should be noted, however, that benign tumors can
produce more than localized lumps, and sometimes they are responsible for serious disease.
Malignant tumors are collectively referred to as cancers, derived from the Latin word for crab, because they
adhere to any part that they seize on in an obstinate manner, similar to a crab. Malignant, as applied to a
neoplasm, implies that the lesion can invade and destroy adjacent structures and spread to distant sites
(metastasize) to cause death. Not all cancers pursue so deadly a course. Some are discovered early and are
treated successfully, but the designation malignant always raises a red flag.
All tumors, benign and malignant, have two basic components: (1) clonal neoplastic cells that constitute
their parenchyma and (2) reactive stroma made up of connective tissue, blood vessels, and variable numbers
of macrophages and lymphocytes. Although the neoplastic cells largely determine a tumor's behavior and
pathologic consequences, their growth and evolution is critically dependent on their stroma. An adequate
stromal blood supply is requisite for the tumor cells to live and divide, and the stromal connective tissue
provides the structural framework essential for the growing cells. In addition, there is cross-talk between
tumor cells and stromal cells that directly influences the growth of tumors. In some tumors, the stromal
support is scant and so the neoplasm is soft and fleshy. In other cases the parenchymal cells stimulate the
formation of an abundant collagenous stroma, referred to as desmoplasia. Some demoplastic tumors—for
example, some cancers of the female breast—are stony hard or scirrhous. The nomenclature of tumors and
their biologic behavior are based primarily on the parenchymal component.

Benign Tumors.
In general, benign tumors are designated by attaching the suffix -oma to the cell of origin. Tumors of
mesenchymal cells generally follow this rule. For example, a benign tumor arising in fibrous tisssue is called
a fibroma, whereas a benign cartilaginous tumor is a chondroma. In contrast, the nomenclature of benign
epithelial tumors is more complex. These are variously classified, some based on their cells of origin, others
on microscopic pattern, and still others on their macroscopic architecture.
Adenoma is applied to a benign epithelial neoplasm derived from glands, although they may or may not
form glandular structures. On this basis, a benign epithelial neoplasm that arises from renal tubular cells
growing in the form of numerous tightly clustered small glands would be termed an adenoma, as would a
heterogeneous mass of adrenal cortical cells growing as a solid sheet. Benign epithelial neoplasms
producing microscopically or macroscopically visible finger-like or warty projections from epithelial
surfaces are referred to as papillomas. Those that form large cystic masses, as in the ovary, are referred to as
cystadenomas. Some tumors produce papillary patterns that protrude into cystic spaces and are called
papillary cystadenomas. When a neoplasm, benign or malignant, produces a macroscopically visible
projection above a mucosal surface and projects, for example, into the gastric or colonic lumen, it is termed
a polyp
Malignant Tumors.
The nomenclature of malignant tumors essentially follows the same schema used for benign neoplasms, with
certain additions. Malignant tumors arising in mesenchymal tissue are usually called sarcomas (Greek sar =
fleshy), because they have little connective tissue stroma and so are fleshy (e.g., fibrosarcoma,
chondrosarcoma, leiomyosarcoma, and rhabdomyosarcoma). Malignant neoplasms of epithelial cell origin,
derived from any of the three germ layers, are called carcinomas. Thus, cancer arising in the epidermis of
ectodermal origin is a carcinoma, as is a cancer arising in the mesodermally derived cells of the renal tubules
and the endodermally derived cells of the lining of the gastrointestinal tract. Carcinomas may be further
qualified. Squamous cell carcinoma would denote a cancer in which the tumor cells resemble stratified
squamous epithelium, and adenocarcinoma denotes a lesion in which the neoplastic epithelial cells grow in
glandular patterns. Sometimes the tissue or organ of origin can be identified, as in the designation of renal
cell adenocarcinoma or bronchogenic squamous cell carcinoma. Not infrequently, however, a cancer is
composed of undifferentiated cells of unknown tissue origin, and must be designated merely as an
undifferentiated malignant tumor.
In many benign and malignant neoplasms, the parenchymal cells bear a close resemblance to each other, as
though all were derived from a single cell. Indeed, neoplasms are of monoclonal origin, as is documented
later. Infrequently, divergent differentiation of a single neoplastic clone along two lineages creates what are
called mixed tumors. The best example of this is the mixed tumor of salivary gland origin. These tumors
contain epithelial components scattered within a myxoid stroma that sometimes contains islands of cartilage
or bone . All these elements, it is believed, arise from a single clone capable of giving rise to epithelial and
myoepithelial cells; thus, the preferred designation of these neoplasms is pleomorphic adenoma. The great
majority of neoplasms, even mixed tumors, are composed of cells representative of a single germ layer. The
multifaceted mixed tumors should not be confused with a teratoma, which contains recognizable mature or
immature cells or tissues representative of more than one germ cell layer and sometimes all three. Teratomas
originate from totipotential cells such as those normally present in the ovary and testis and sometimes
abnormally present in sequestered midline embryonic rests. Such cells have the capacity to differentiate into
any of the cell types found in the adult body and so, not surprisingly, may give rise to neoplasms that mimic,
in a helter-skelter fashion, bits of bone, epithelium, muscle, fat, nerve, and other tissues. When all the
component parts are well differentiated, it is a benign (mature) teratoma; when less well differentiated, it is
an immature, potentially or overtly, malignant teratoma. A particularly common pattern is seen in the
ovarian cystic teratoma (dermoid cyst), which differentiates principally along ectodermal lines to create a
cystic tumor lined by skin replete with hair, sebaceous glands, and tooth structures.

The nomenclature of the more common forms of neoplasia is presented in Table 7-1 . It is evident from this
compilation that there are some inappropriate but deeply entrenched usages. For generations, benign-
sounding designations such as lymphoma, melanoma, mesothelioma, and seminoma have been used for
certain malignant neoplasms. The converse is also true; ominous terms may be applied to trivial lesions.
Hamartomas present as disorganized but benign-appearing masses composed of cells indigenous to the
particular site. They were once thought to be a developmental malformation, unworthy of the -oma
designation. For example, pulmonary chondroid harmatoma contains islands of disorganized, but
histologically normal cartilage, bronchi, and vessels. However, many hamartomas, including pulmonary
chondroid hamartoma, have clonal, recurrent translocations involving genes encoding certain chromatin
proteins.
[3]
Thus, through molecular biology they have finally earned their -oma designation. Another
misnomer is the term choristoma. This congenital anomaly is better described as a heterotopic rest of cells.
For example, a small nodule of well-developed and normally organized pancreatic substance may be found
in the submucosa of the stomach, duodenum, or small intestine. This heterotopic rest may be replete with
islets of Langerhans and exocrine glands. The term choristoma, connoting a neoplasm, imparts to the
heterotopic rest a gravity far beyond its usual trivial significance. Although regrettably the terminology of
neoplasms is not simple, it is important because it is the language by which the nature and significance of
tumors are categorized.
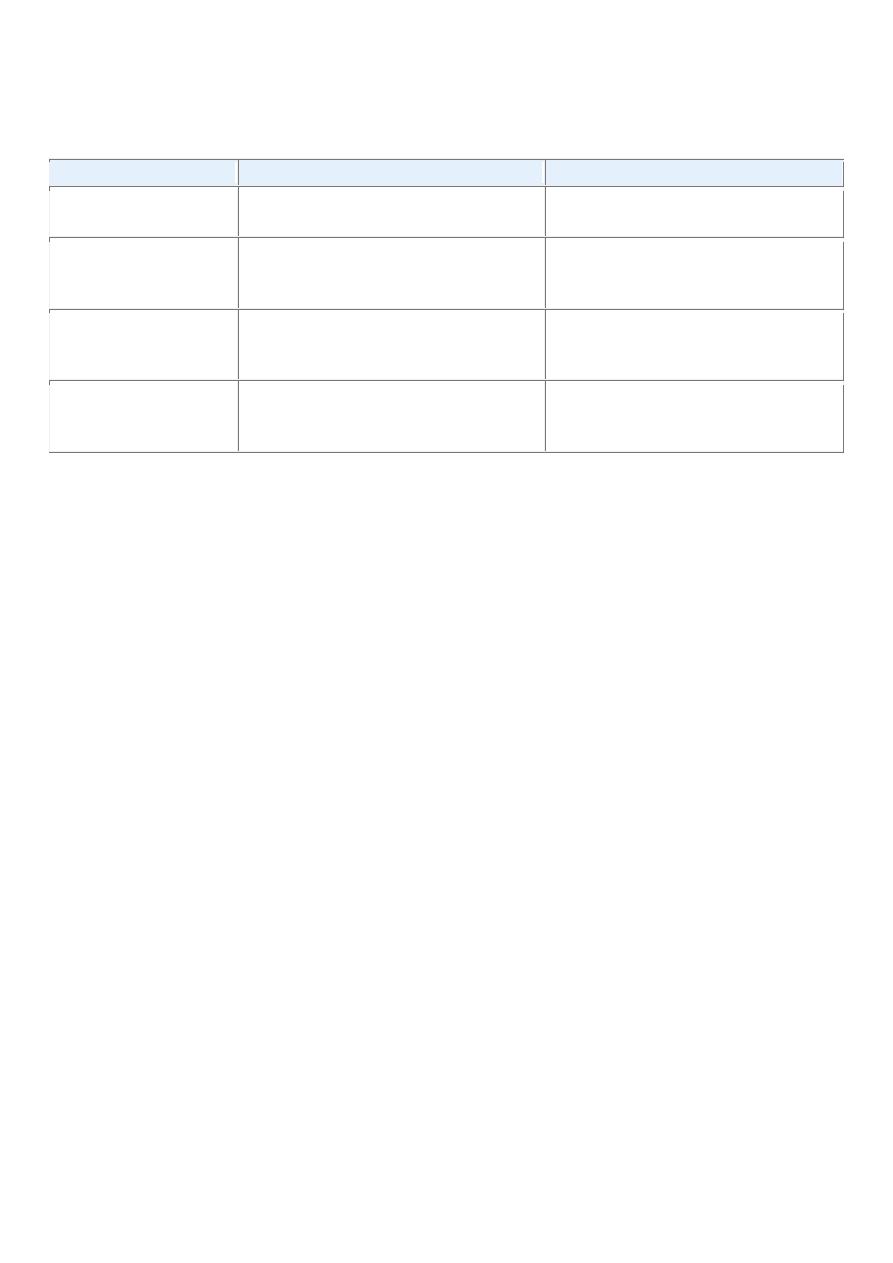
TABLE 7-1 -- Nomenclature of Tumors
Tissue of Origin
Benign
Malignant
COMPOSED OF ONE PARENCHYMAL CELL TYPE
Tumors of Mesenchymal Origin
Connective tissue and derivatives Fibroma
Fibrosarcoma
Lipoma
Liposarcoma
Chondroma
Chondrosarcoma
Osteoma
Osteogenic sarcoma
Endothelial and Related Tissues
Blood vessels
Hemangioma
Angiosarcoma
Lymph vessels
Lymphangioma
Lymphangiosarcoma
Synovium
Synovial sarcoma
Mesothelium
Mesothelioma
Brain coverings
Meningioma
Invasive meningioma
Blood Cells and Related Cells
Hematopoietic cells
Leukemias
Lymphoid tissue
Lymphomas
Muscle
Smooth
Leiomyoma
Leiomyosarcoma
Striated
Rhabdomyoma
Rhabdomyosarcoma
Tumors of Epithelial Origin
Stratified squamous
Squamous cell papilloma
Squamous cell carcinoma
Basal cells of skin or adnexa
Basal cell carcinoma
Epithelial lining of glands or
ducts
Adenoma
Adenocarcinoma
Papilloma
Papillary carcinomas

Tissue of Origin
Benign
Malignant
Cystadenoma
Cystadenocarcinoma
Respiratory passages
Bronchial adenoma
Bronchogenic carcinoma
Renal epithelium
Renal tubular adenoma
Renal cell carcinoma
Liver cells
Liver cell adenoma
Hepatocellular carcinoma
Urinary tract epithelium
(transitional)
Transitional-cell papilloma
Transitional-cell carcinoma
Placental epithelium
Hydatidiform mole
Choriocarcinoma
Testicular epithelium (germ
cells)
Seminoma
Embryonal carcinoma
Tumors of Melanocytes
Nevus
Malignant melanoma
MORE THAN ONE NEOPLASTIC CELL TYPE—MIXED TUMORS, USUALLY DERIVED
FROM ONE GERM CELL LAYER
Salivary glands
Pleomorphic adenoma (mixed tumor
of salivary origin)
Malignant mixed tumor of salivary
gland origin
Renal anlage
Wilms tumor
MORE THAN ONE NEOPLASTIC CELL TYPE DERIVED FROM MORE THAN ONE GERM
CELL LAYER—TERATOGENOUS
Totipotential cells in gonads or in
embryonic rests
Mature teratoma, dermoid cyst
Immature teratoma,
teratocarcinoma
Characteristics of Benign and Malignant Neoplasms
DIFFERENTIATION AND ANAPLASIA
Differentiation refers to the extent to which neoplastic parenchymal cells resemble the corresponding
normal parenchymal cells, both morphologically and functionally; lack of differentiation is called
anaplasia. In general, benign tumors are well differentiated ( Figs. 7-4 and 7-5 ). The neoplastic cell in a
benign adipocyte tumor—a lipoma—so closely resembles the normal cell that it may be impossible to
recognize it as a tumor by microscopic examination of individual cells. Only the growth of these cells into a
discrete mass discloses the neoplastic nature of the lesion. One may get so close to the tree that one loses
sight of the forest. In well-differentiated benign tumors, mitoses are extremely scant in number and are of
normal configuration.
Malignant neoplasms are characterized by a wide range of parenchymal cell differentiation, from
surprisingly well differentiated to completely undifferentiated. Certain well-differentiated adenocarcinomas
of the thyroid, for example, may form normal-appearing follicles, and some squamous cell carcinomas
contain cells that do not differ cytologically from normal squamous epithelial cells.
Thus, the morphologic diagnosis of malignancy in well-differentiated tumors may sometimes be quite
difficult. In between the two extremes lie tumors that are loosely referred to as moderately well
differentiated.

Neoplasms that are composed of poorly differentiated cells are said to be anaplastic. Lack of differentiation,
or anaplasia, is considered a hallmark of malignancy. The term anaplasia literally means “to form
backward,” implying a reversal of differentiation to a more primitive level. It is believed, however, that most
cancers do not represent “reverse differentiation” of mature normal cells but, in fact, arise from less mature
cells with “stem-cell-like” properties, such as tissue stem cells ( Chapter 3 ). In well-differentiated tumors,
daughter cells derived from these “cancer stem cells” retain the capacity for differentiation, whereas in
poorly differentiated tumors that capacity is lost.
Lack of differentiation, or anaplasia, is often associated with many other morphologic changes.
• Pleomorphism. Both the cells and the nuclei characteristically display pleomorphism—variation in
size and shape . Thus, cells within the same tumor are not uniform, but range from large cells, many
times larger than their neighbors, to extremely small and primitive appearing.
• Abnormal nuclear morphology. Characteristically the nuclei contain abundant chromatin and are
dark staining (hyperchromatic). The nuclei are disproportionately large for the cell, and the nuclear-
to-cytoplasm ratio may approach 1 : 1 instead of the normal 1 : 4 or 1 : 6. The nuclear shape is
variable and often irregular, and the chromatin is often coarsely clumped and distributed along the
nuclear membrane. Large nucleoli are usually present in these nuclei.
• Mitoses. As compared with benign tumors and some well-differentiated malignant neoplasms,
undifferentiated tumors usually possess large numbers of mitoses, reflecting the higher proliferative
activity of the parenchymal cells. The presence of mitoses, however, does not necessarily indicate
that a tumor is malignant or that the tissue is neoplastic. Many normal tissues exhibiting rapid
turnover, such as bone marrow, have numerous mitoses, and non-neoplastic proliferations such as
hyperplasias contain many cells in mitosis. More important as a morphologic feature of malignancy
are atypical, bizarre mitotic figures, sometimes producing tripolar, quadripolar, or multipolar
spindles.
• Loss of polarity. In addition to the cytologic abnormalities, the orientation of anaplastic cells is
markedly disturbed (i.e., they lose normal polarity). Sheets or large masses of tumor cells grow in an
anarchic, disorganized fashion.
• Other changes. Another feature of anaplasia is the formation of tumor giant cells, some possessing
only a single huge polymorphic nucleus and others having two or more large, hyperchromatic nuclei.
These giant cells are not to be confused with inflammatory Langhans or foreign body giant cells,
which are derived from macrophages and contain many small, normal-appearing nuclei. Although
growing tumor cells obviously require a blood supply, often the vascular stroma is scant, and in
many anaplastic tumors, large central areas undergo ischemic necrosis.
Before we leave the subject of differentiation and anaplasia, we should discuss metaplasia and dysplasia.
Metaplasia is defined as the replacement of one type of cell with another type. Metaplasia is nearly always
found in association with tissue damage, repair, and regeneration. Often the replacing cell type is more
suited to a change in environment. For example, gastroesophageal reflux damages the squamous epithelium
of the esophagus, leading to its replacement by glandular (gastric or intestinal) epithelium, more suited to the
acidic environment. Dysplasia is a term that literally means disordered growth. Dysplasia often occurs in
metaplastic epithelium, but not all metaplastic epithelium is also dysplastic. Dysplasia is encountered
principally in epithelia, and it is characterized by a constellation of changes that include a loss in the
uniformity of the individual cells as well as a loss in their architectural orientation. Dysplastic cells exhibit
considerable pleomorphism and often contain large hyperchromatic nuclei with a high nuclearto-
cytoplasmic ratio. The architecture of the tissue may be disorderly. For example, in squamous epithelium the
usual progressive maturation of tall cells in the basal layer to flattened squames on the surface may be lost
and replaced by a scrambling of dark basal-appearing cells throughout the epithelium. Mitotic figures are
more abundant than usual, although almost invariably they have a normal configuration. Frequently,

however, the mitoses appear in abnormal locations within the epithelium. For example, in dysplastic
stratified squamous epithelium, mitoses are not confined to the basal layers but instead may appear at all
levels, including surface cells. When dysplastic changes are marked and involve the entire thickness of the
epithelium but the lesion remains confined by the basement membrane, it is considered a preinvasive
neoplasm and is referred to as carcinoma in situ. Once the tumor cells breach the basement membrane, the
tumor is said to be invasive. Dysplastic changes are often found adjacent to foci of invasive carcinoma, and
in some situations, such as in long-term cigarette smokers and persons with Barrett esophagus, severe
epithelial dysplasia frequently antedates the appearance of cancer. However, dysplasia does not necessarily
progress to cancer. Mild to moderate changes that do not involve the entire thickness of epithelium may be
reversible, and with removal of the inciting causes the epithelium may revert to normal. Even carcinoma in
situ may take years to become invasive.
. Despite exceptions, the more rapidly growing and the more anaplastic a tumor, the less likely it will have
specialized functional activity. The cells in benign tumors are almost always well differentiated and
resemble their normal cells of origin; the cells in cancer are more or less differentiated, but some
derangement of differentiation is always present.
RATES OF GROWTH
A fundamental issue in tumor biology is to understand the factors that affect the growth rates of tumors and
their influence on clinical outcome and therapeutic responses. One can begin the consideration of tumor cell
kinetics by asking the question: How long does it take to produce a clinically overt tumor mass? It is a
reasonable estimate the original transformed cell (approximately 10 μm in diameter) must undergo at least
30 population doublings to produce 10
9
cells (weighing approximately 1 gm), which is the smallest
clinically detectable mass. In contrast, only 10 additional doubling cycles are required to produce a tumor
containing 10
12
cells (weighing -1kg), which is usually the maximal size compatible with life. These are
minimal estimates, based on the assumption that all descendants of the transformed cell retain the ability to
divide and that there is no loss of cells from the replicative pool. This concept of tumor as a “pathologic
dynamo” is not entirely correct, as we discuss subsequently. Nevertheless, this calculation highlights an
extremely important concept about tumor growth: By the time a solid tumor is clinically detected, it has
already completed a major portion of its life span. This is a major impediment in the treatment of cancer and
underscores the need to develop diagnostic markers to detect early cancers.
The rate of growth of a tumor is determined by three main factors: the doubling time of tumor cells, the
fraction of tumor cells that are in the replicative pool, and the rate at which cells are shed or die. Because
cell cycle controls are deranged in most tumors, tumor cells can be triggered to cycle without the usual
restraints. The dividing cells, however, do not necessarily complete the cell cycle more rapidly than do
normal cells. In reality, total cell cycle time for many tumors is equal to or longer than that of corresponding
normal cells. Thus, it can be safely concluded that growth of tumors is not commonly associated with a
shortening of cell cycle time.
The proportion of cells within the tumor population that are in the proliferative pool is referred to as the
growth fraction. Clinical and experimental studies suggest that during the early, submicroscopic phase of
tumor growth, the vast majority of transformed cells are in the proliferative pool . As tumors continue to
grow, cells leave the proliferative pool in ever-increasing numbers as a result of shedding, lack of nutrients,
necrosis, apoptosis, differentiation, and reversion to the nonproliferative phase of the cell cycle (G
0
). Thus,
by the time a tumor is clinically detectable, most cells are not in the replicative pool. Even in some rapidly
growing tumors, the growth fraction is only about 20% or less.
Ultimately the progressive growth of tumors and the rate at which they grow are determined by an excess of
cell production over cell loss. In some tumors, especially those with a relatively high growth fraction, the
imbalance is large, resulting in more rapid growth than in those in which cell production exceeds cell loss by
only a small margin. Some leukemias and lymphomas and certain lung cancers (i.e., small-cell carcinoma)

have a relatively high growth fraction, and their clinical course is rapid. By comparison, many common
tumors, such as cancers of the colon and breast, have low growth fractions, and cell production exceeds cell
loss by only about 10%; they tend to grow at a much slower pace.
Several important conceptual and practical lessons can be learned from studies of tumor cell kinetics:
• Fast-growing tumors may have a high cell turnover, implying that rates of both proliferation and
apoptosis are high. Obviously if the tumor is to grow, the rate of proliferation must exceed that of
cell death.
• The growth fraction of tumor cells has a profound effect on their susceptibility to cancer
chemotherapy. Because most anticancer agents act on cells that are in cycle, it is not difficult to
imagine that a tumor that contains 5% of all cells in the replicative pool will be slow growing but
relatively refractory to treatment with drugs that kill dividing cells. One strategy used in the
treatment of tumors with low growth fraction (e.g., cancer of colon and breast) is first to shift tumor
cells from G
0
into the cell cycle. This can be accomplished by debulking the tumor with surgery or
radiation. The surviving tumor cells tend to enter the cell cycle and thus become susceptible to drug
therapy. Such considerations form the basis of combined-modality treatment. Some aggressive
tumors (such as certain lymphomas and leukemias) that contain a large pool of dividing cells literally
melt away with chemotherapy and may even be cured.
We can now return to the question posed earlier: How long does it take for one transformed cell to produce a
clinically detectable tumor containing 10
9
cells? If every one of the daughter cells remained in cell cycle and
no cells were shed or lost, we could anticipate the answer to be 90 days (30 population doublings, with a cell
cycle time of 3 days). In reality, the latent period before which a tumor becomes clinically detectable is
unpredictable but typically much longer than 90 days, as long as many years for most solid tumors,
emphasizing once again that human cancers are diagnosed only after they are fairly advanced in their life
cycle. After they become clinically detectable, the average volume-doubling time for such common killers as
cancer of the lung and colon is about 2 to 3 months. As might be anticipated from the discussion of the
variables that affect growth rate, however, the range of doubling time values is extremely broad, varying
from less than 1 month for some childhood cancers to more than 1 year for certain salivary gland tumors.
Cancer is indeed an unpredictable group of disorders.
In general, the growth rate of tumors correlates with their level of differentiation, and thus most malignant
tumors grow more rapidly than do benign lesions. There are, however, many exceptions to such an
oversimplification. Some benign tumors have a higher growth rate than malignant tumors. Moreover, the
rate of growth of benign as well as malignant neoplasms may not be constant over time. Factors such as
hormonal stimulation, adequacy of blood supply, and unknown influences may affect their growth. For
example, the growth of uterine leiomyomas (benign smooth muscle tumors) may change over time because
of hormonal variations. Not infrequently, repeated clinical examination of women bearing such neoplasms
over the span of decades discloses no significant increase in size. After menopause the neoplasms may
atrophy and may be replaced largely by collagenous, sometimes calcified, tissue. During pregnancy
leiomyomas frequently enter a growth spurt. Such changes reflect the responsiveness of the tumor cells to
circulating levels of steroid hormones, particularly estrogens. Cancers show a wide range of growth. Some
malignant tumors grow slowly for years and then suddenly increase in size, explosively disseminating to
cause death within a few months of discovery. It is possible that such behavior results from the emergence
of an aggressive subclone of transformed cells. At the other extreme are malignant neoplasms that grow
more slowly than do benign tumors and may even enter periods of dormancy lasting for years. On occasion,
cancers decrease in size and even spontaneously disappear, but such “miracles” are rare enough that they
remain intriguing curiosities.

CANCER STEM CELLS AND CANCER CELL LINEAGES
Cancers are immortal and have limitless proliferative capacity, indicating that like normal tissues, they also
must contain cells with “stemlike” properties. The concept of cancer stem cells has several important
implications. Most notably, if cancer stem cells are essential for tumor persistence, it follows that these cells
must be eliminated to cure the affected patient. It is hypothesized that like normal stem cells, cancer stem
cells have a high intrinsic resistance to conventional therapies, because of their low rate of cell division and
the expression of factors, such as multiple drug resistance-1 (MDR1), that counteract the effects of
chemotherapeutic drugs.Thus, the limited success of current therapies may in part be explained by their
failure to kill the malignant stem cells that lie at the root of cancer. Cancer stem cells could arise from
normal tissue stem cells or from more differentiated cells that, as part of the transformation process, acquire
the property of self-renewal. Studies of certain leukemias support both of these possibilities. For example,
chronic myelogenous leukemia (CML) originates from the malignant counterpart of a normal hematopoietic
stem cell, whereas certain acute myeloid leukemias (AMLs) are derived from more differentiated myeloid
precursors that acquire an abnormal capacity for self-renewal. The identification of “leukemia stem cells”
has spurred the search for cancer stem cells in solid tumors. Most such studies have focused on the
identification of tumor-initiating cells (T-ICs), which are defined as cells that allow a human tumor to grow
and maintain itself indefinitely when transplanted into an immunodeficient mouse. T-ICs have been
identified in several human tumors, including breast carcinoma, glioblastoma multiforme, colon cancer, and
AML, in which they constitute 0.1% to 2% of the total cellularity.
LOCAL INVASION
Nearly all benign tumors grow as cohesive expansile masses that remain localized to their site of origin and
do not have the capacity to infiltrate, invade, or metastasize to distant sites, as do malignant tumors. Because
they grow and expand slowly, they usually develop a rim of compressed connective tissue, sometimes called
a fibrous capsule, which separates them from the host tissue. This capsule is derived largely from the
extracellular matrix of the native tissue due to atrophy of normal parenchymal cells under the pressure of an
expanding tumor. Such encapsulation does not prevent tumor growth, but it keeps the benign neoplasm as a
discrete, readily palpable, and easily movable mass that can be surgically enucleated. Although a well-
defined cleavage plane exists around most benign tumors, in some it is lacking. For example, hemangiomas
(neoplasms composed of tangled blood vessels) are often unencapsulated and may appear to permeate the
site in which they arise (commonly the dermis of the skin).
The growth of cancers is accompanied by progressive infiltration, invasion, and destruction of the
surrounding tissue. In general, malignant tumors are poorly demarcated from the surrounding normal tissue,
and a well-defined cleavage plane is lacking. Slowly expanding malignant tumors, however, may develop an
apparently enclosing fibrous capsule and may push along a broad front into adjacent normal structures.
Histologic examination of such pseudo-encapsulated masses almost always shows rows of cells penetrating
the margin and infiltrating the adjacent structures, a crablike pattern of growth that constitutes the popular
image of cancer.
Most malignant tumors are obviously invasive and can be expected to penetrate the wall of the colon or
uterus, for example, or fungate through the surface of the skin. They recognize no normal anatomic
boundaries. Such invasiveness makes their surgical resection difficult or impossible, and even if the tumor
appears well circumscribed it is necessary to remove a considerable margin of apparently normal tissues
adjacent to the infiltrative neoplasm. Next to the development of metastases, invasiveness is the most reliable
feature that differentiates malignant from benign tumors. We noted earlier that some cancers seem to evolve
from a preinvasive stage referred to as carcinoma in situ. This commonly occurs in carcinomas of the skin,
breast, and certain other sites and is best illustrated by carcinoma of the uterine cervix ( Chapter 22 ). In situ

epithelial cancers display the cytologic features of malignancy without invasion of the basement membrane.
They may be considered one step removed from invasive cancer; with time, most penetrate the basement
membrane and invade the subepithelial stroma.
METASTASIS
Metastases are tumor implants discontinuous with the primary tumor. Metastasis unequivocally marks a
tumor as malignant because benign neoplasms do not metastasize. The invasiveness of cancers permits them
to penetrate into blood vessels, lymphatics, and body cavities, providing the opportunity for spread. With few
exceptions, all malignant tumors can metastasize. The major exceptions are most malignant neoplasms of
the glial cells in the central nervous system, called gliomas, and basal cell carcinomas of the skin. Both are
locally invasive forms of cancer, but they rarely metastasize. It is evident then that the properties of invasion
and metastasis are separable.
In general, the more aggressive, the more rapidly growing, and the larger the primary neoplasm, the greater
the likelihood that it will metastasize or already has metastasized. There are innumerable exceptions,
however. Small, well-differentiated, slowly growing lesions sometimes metastasize widely; conversely,
some rapidly growing, large lesions remain localized for years. Many factors relating to both invader and
host are involved.
Approximately 30% of newly diagnosed individuals with solid tumors (excluding skin cancers other than
melanomas) present with metastases. Metastatic spread strongly reduces the possibility of cure; hence, short
of prevention of cancer, no achievement would be of greater benefit to patients than methods to block
metastases.
Pathways of Spread
Dissemination of cancers may occur through one of three pathways: (1) direct seeding of body cavities or
surfaces, (2) lymphatic spread, and (3) hematogenous spread. Although direct transplantation of tumor cells,
as for example on surgical instruments, may theoretically occur, it is rare and we do not discuss this artificial
mode of dissemination further. Each of the three major pathways is described separately.
Seeding of Body Cavities and Surfaces.
Seeding of body cavities and surfaces may occur whenever a malignant neoplasm penetrates into a natural
“open field.” Most often involved is the peritoneal cavity , but any other cavity—pleural, pericardial,
subarachnoid, and joint space—may be affected. Such seeding is particularly characteristic of carcinomas
arising in the ovaries, when, not infrequently, all peritoneal surfaces become coated with a heavy layer of
cancerous glaze. Remarkably, the tumor cells may remain confined to the surface of the coated abdominal
viscera without penetrating into the substance. Sometimes mucus-secreting appendiceal carcinomas fill the
peritoneal cavity with a gelatinous neoplastic mass referred to as pseudomyxoma peritonei.
Lymphatic Spread.
Transport through lymphatics is the most common pathway for the initial dissemination of carcinomas, and
sarcomas may also use this route. Tumors do not contain functional lymphatics, but lymphatic vessels
located at the tumor margins are apparently sufficient for the lymphatic spread of tumor cells.The emphasis
on lymphatic spread for carcinomas and hematogenous spread for sarcomas is misleading, because
ultimately there are numerous interconnections between the vascular and the lymphatic systems. The pattern
of lymph node involvement follows the natural routes of lymphatic drainage. Because carcinomas of the
breast usually arise in the upper outer quadrants, they generally disseminate first to the axillary lymph nodes.
Cancers of the inner quadrants drain to the nodes along the internal mammary arteries. Thereafter the
infraclavicular and supraclavicular nodes may become involved. Carcinomas of the lung arising in the major

respiratory passages metastasize first to the perihilar tracheobronchial and mediastinal nodes. Local lymph
nodes, however, may be bypassed—so-called “skip metastasis”—because of venous-lymphatic anastomoses
or because inflammation or radiation has obliterated lymphatic channels.
In breast cancer, determining the involvement of axillary lymph nodes is very important for assessing the
future course of the disease and for selecting suitable therapeutic strategies. To avoid the considerable
surgical morbidity associated with a full axillary lymph node dissection, biopsy of sentinel nodes is often
used to assess the presence or absence of metastatic lesions in the lymph nodes. A sentinel lymph node is
defined as “the first node in a regional lymphatic basin that receives lymph flow from the primary
tumor.”Sentinel node mapping can be done by injection of radiolabeled tracers and blue dyes, and the use of
frozen section upon the sentinel lymph node at the time of surgery can guide the surgeon to the appropriate
therapy. Sentinel node biopsy has also been used for detecting the spread of melanomas, colon cancers, and
other tumors.
In many cases the regional nodes serve as effective barriers to further dissemination of the tumor, at least for
a while. Conceivably the cells, after arrest within the node, may be destroyed by a tumor-specific immune
response. Drainage of tumor cell debris or tumor antigens, or both, also induces reactive changes within
nodes. Thus, enlargement of nodes may be caused by (1) the spread and growth of cancer cells or (2)
reactive hyperplasia . Therefore, nodal enlargement in proximity to a cancer, while it must arouse suspicion,
does not necessarily mean dissemination of the primary lesion.
Hematogenous Spread.
Hematogenous spread is typical of sarcomas but is also seen with carcinomas. Arteries, with their thicker
walls, are less readily penetrated than are veins. Arterial spread may occur, however, when tumor cells pass
through the pulmonary capillary beds or pulmonary arteriovenous shunts or when pulmonary metastases
themselves give rise to additional tumor emboli. In such vascular spread, several factors influence the
patterns of distribution of the metastases. With venous invasion the blood-borne cells follow the venous flow
draining the site of the neoplasm, and the tumor cells often come to rest in the first capillary bed they
encounter. Understandably the liver and lungs are most frequently involved in such hematogenous
dissemination ( Figs. 7-18 and 7-19 ), because all portal area drainage flows to the liver and all caval blood
flows to the lungs. Cancers arising in close proximity to the vertebral column often embolize through the
paravertebral plexus, and this pathway is involved in the frequent vertebral metastases of carcinomas of the
thyroid and prostate.
Certain cancers have a propensity for invasion of veins. Renal cell carcinoma often invades the branches of
the renal vein and then the renal vein itself to grow in a snakelike fashion up the inferior vena cava,
sometimes reaching the right side of the heart. Hepatocellular carcinomas often penetrate portal and hepatic
radicles to grow within them into the main venous channels. Remarkably, such intravenous growth may not
be accompanied by widespread dissemination. Histologic evidence of penetration of small vessels at the site
of the primary neoplasm is obviously an ominous feature. Such changes, however, must be viewed
guardedly because, for reasons discussed later, they do not indicate the inevitable development of
metastases.
Many observations suggest that mere anatomic localization of the neoplasm and natural pathways of venous
drainage do not wholly explain the systemic distributions of metastases. For example, breast carcinoma
preferentially spreads to bone, bronchogenic carcinomas tend to involve the adrenals and the brain, and
neuroblastomas spread to the liver and bones. Conversely, skeletal muscles and the spleen, despite the large
percentage of blood flow they receive and the enormous vascular beds present, are rarely the site of
secondary deposits. The probable basis of such tissue-specific homing of tumor cells is discussed later.
The distinguishing features of benign and malignant tumors discussed in this overview are summarized in
Table 7-2. With this background on the structure and behavior of neoplasms, we now discuss the origin of
tumors, starting with insights gained from the epidemiology of cancer and followed by the molecular basis
of carcinogenesis.
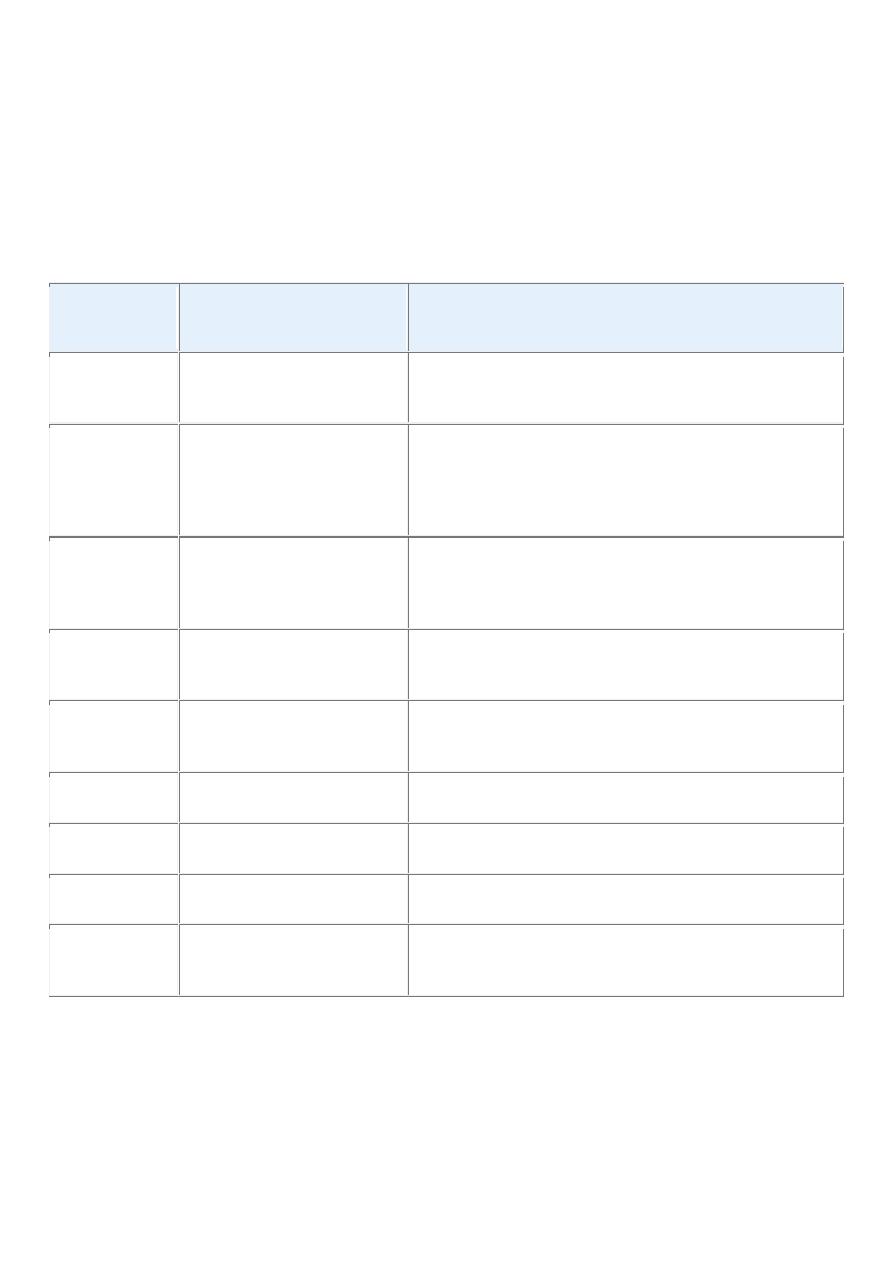
TABLE 7-2 -- Comparisons between Benign and Malignant Tumors
Characteristics
Benign
Malignant
Differentiation/anaplasia Well differentiated; structure sometimes
typical of tissue of origin
Some lack of differentiation with
anaplasia; structure often atypical
Rate of growth
Usually progressive and slow; may come
to a standstill or regress; mitotic figures
rare and normal
Erratic and may be slow to rapid;
mitotic figures may be numerous and
abnormal
Local invasion
Usually cohesive expansile well-
demarcated masses that do not invade or
infiltrate surrounding normal tissues
Locally invasive, infiltrating
surrounding tissue; sometimes may be
seemingly cohesive and expansile
Metastasis
Absent
Frequently present; the larger and more
undifferentiated the primary, the more
likely are metastases
Epidemiology
Because cancer is a disorder of cell growth and behavior, its ultimate cause has to be defined at the cellular
and subcellular levels. Study of cancer patterns in populations, however, can contribute substantially to
knowledge about the origins of cancer. Epidemiologic studies have established the causative link between
smoking and lung cancer, and comparison of diet and cancer rates in the Western world and Africa has
implicated high dietary fat and low fiber in the development of colon cancer. Major insights into the causes
of cancer can be obtained by epidemiologic studies that relate particular environmental, racial (possibly
hereditary), and cultural influences to the occurrence of specific neoplasms. Certain diseases associated with
an increased risk of developing cancer (preneoplastic disorders) also provide clues to the pathogenesis of
cancer. In the following discussion we first summarize the overall incidence of cancer to gain an insight into
the magnitude of the cancer problem, then we review some factors relating to the patient and environment
that influence the predisposition to cancer.
CANCER INCIDENCE
In some measure, an individual's likelihood of developing a cancer is expressed by national incidence and
mortality rates. For example, residents of the United States have about a one in five chance of dying of
cancer. There were, it is estimated, about 1,437,180 new cancer cases and 565,650 deaths from cancer in
2008, representing 23% of all mortality,
[1]
a frequency surpassed only by deaths caused by cardiovascular
diseases. These data do not include an additional 1 million, for the most part readily curable, non-melanoma
cancers of the skin and 122,000 cases of carcinoma in situ, largely of the female breast and melanomas.
[1]
The major organ sites affected and the estimated frequency of cancer deaths are shown in Figure 7-21 . The
most common tumors in men arise in the prostate, lung, and colorectum. In women, cancers of the breast,
lung, and colon and rectum are the most frequent. Cancers of the lung, female breast, prostate, and
colon/rectum constitute more than 50% of cancer diagnoses and cancer deaths in the U.S. population.
[1]
The age-adjusted death rates (number of deaths per 100,000 population) for many forms of cancer have
significantly changed over the years. Many of the long-term comparisons are noteworthy. Over the last 50
years of the twentieth century, the overall age-adjusted cancer death rate significantly increased in both men

and women. However, since 1995 the cancer incidence rate in men has stabilized and since 1990 the cancer
death rate in men has decreased 18.4%. In women the cancer incidence rate stabilized in 1995, and the
cancer death rate has decreased 10.4% since 1991. Among men nearly 80% of the total decrease in cancer
death rates is accounted for by decreases in death rates from lung, prostate, and colorectal cancers since
1990.
Among women nearly 60% of the decrease in cancer death rates is due to reductions in death rates
from breast and colorectal cancers. Nearly 40% of the sex-specific decreases in cancer death rates is
accounted for by a reduction in lung cancer deaths in men and breast cancer deaths in women.
[1]
Decreased
use of tobacco products is responsible for the reduction in lung cancer deaths, while improved detection and
treatment are responsible for the decrease in death rates for colorectal, female breast, and prostate cancer.
[1]
The last half century has seen a decline in the number of deaths caused by cervical cancer that relates to
earlier diagnosis made possible by the Papanicolaou (Pap) smear. The downward trend in deaths from
stomach cancer has been attributed to a decrease in some dietary carcinogens, as a consequence of better
food preservation or changes in dietary habits. Unfortunately, between 1990–1991 and 2004, lung cancer
death rates in women, and liver and intrahepatic bile duct cancer death rates in men, increased substantially,
offsetting some of the improvement in survival from other cancers. Indeed, although in women carcinomas
of the breast occur about 2.5 times more frequently than those of the lung, lung cancer has become the
leading cause of cancer deaths in women. Deaths from primary liver cancers, which declined between 1930
and 1970, have approximately doubled during the past 30 years. This number is expected to increase over
the coming decades, as the large number of individuals infected with the hepatitis C virus (HCV) begin to
develop hepatocellular carcinoma.
Although race is not a strict biologic category, it can define groups at risk for certain cancers.
The disparity
in cancer mortality rates between white and black Americans persists, but African Americans had the largest
decline in cancer mortality during the past decade. Hispanics living in the United States have a lower
frequency of the most common tumors than the white non-Hispanic population but a higher incidence of
tumors of the stomach, liver, uterine cervix, and gallbladder, as well as certain childhood leukemias.
GEOGRAPHIC AND ENVIRONMENTAL FACTORS
Although genetics and environmental triggers both play a role in the pathogenesis of cancer, environmental
factors are thought to be the more significant contributors in most common sporadic cancers. In one large
study the proportion of risk from environmental causes was found to be 65%, whereas heritable factors
contributed 26% to 42% of cancer risk. Remarkable differences found in the incidence and death rates of
specific forms of cancer around the world also suggest a role for environmental factors. For example, the
death rate for stomach carcinoma in both men and women is seven to eight times higher in Japan than in the
United States. In contrast, the death rate from carcinoma of the lung is slightly more than twice as great in
the United States as in Japan. Although racial predispositions cannot be ruled out, it is generally believed
that most of these geographic differences are the consequence of environmental influences. Indeed,
comparing mortality rates for Japanese immigrants to the United States and Japanese born in the United
States of immigrant parents (Nisei) with those of long-term residents of both countries shows that cancer
mortality rates for first-generation Japanese immigrants are intermediate between those of natives of Japan
and natives of California, and the two rates come closer with each passing generation . This points strongly
to environmental and cultural factors rather than genetic predisposition.
There is no paucity of carcinogenic environmental factors: they lurk in the ambient environment, in the
workplace, in food, and in personal practices. Individuals may be exposed to carcinogenic factors when they
go outside (ultraviolet [UV] rays, smog), in their medication (methotrexate), at work (asbestos, vinyl
chloride; Table 7-3 ), or at home (high-fat diet, alcohol). Overall, mortality data indicate that the most
overweight individuals in the U.S. population have a 52% (men) and 62% (women) higher death rate from
cancer than do their slimmer counterparts. Indeed, obesity is associated with approximately 14% of cancer
deaths in men and 20% in women.Alcohol abuse alone increases the risk of carcinomas of the oropharynx
(excluding lip), larynx, and esophagus and, by the development of alcoholic cirrhosis, hepatocellular
carcinoma. Smoking, particularly of cigarettes, has been implicated in cancer of the mouth, pharynx, larynx,
esophagus, pancreas, bladder, and most significantly, about 90% of lung cancer deaths. Cigarette smoking
has been called the single most important environmental factor contributing to premature death in the United
States. Alcohol and tobacco together synergistically increase the danger of incurring cancers in the upper
aerodigestive tract. The risk of cervical cancer is linked to age at first intercourse and the number of sex
partners, and it is now known that infection by venereally transmitted human papillomavirus (HPV)
contributes to cervical dysplasia and cancer. It appears that almost everything one does to gain a livelihood
or for pleasure is fattening, immoral, illegal, or, even worse, oncogenic.
TABLE 7-3 -- Occupational Cancers
Agents or
Groups of
Agents
Human Cancer Site for
Which Reasonable Evidence
Is Available
Typical Use or Occurrence
Arsenic and
arsenic
compounds
Lung, skin, hemangiosarcoma Byproduct of metal smelting; component of alloys,
electrical and semiconductor devices, medications and
herbicides, fungicides, and animal dips
Asbestos
Lung, mesothelioma;
gastrointestinal tract
(esophagus, stomach, large
intestine)
Formerly used for many applications because of fire, heat,
and friction resistance; still found in existing construction
as well as fire-resistant textiles, friction materials (i.e.,
brake linings), underlayment and roofing papers, and floor
tiles
Benzene
Leukemia, Hodgkin
lymphoma
Principal component of light oil; despite known risk, many
applications exist in printing and lithography, paint,
rubber, dry cleaning, adhesives and coatings, and
detergents; formerly widely used as solvent and fumigant
Beryllium and
beryllium
compounds
Lung
Missile fuel and space vehicles; hardener for lightweight
metal alloys, particularly in aerospace applications and
nuclear reactors
Cadmium and
cadmium
compounds
Prostate
Uses include yellow pigments and phosphors; found in
solders; used in batteries and as alloy and in metal platings
and coatings
Chromium
compounds
Lung
Component of metal alloys, paints, pigments, and
preservatives
Nickel
compounds
Nose, lung
Nickel plating; component of ferrous alloys, ceramics, and
batteries; by-product of stainless-steel arc welding
Radon and its
decay products
Lung
From decay of minerals containing uranium; potentially
serious hazard in quarries and underground mines
Vinyl chloride
Angiosarcoma, liver
Refrigerant; monomer for vinyl polymers; adhesive for
plastics; formerly inert aerosol propellant in pressurized
containers
Modified from Stellman JM, Stellman SD: Cancer and workplace. CA Cancer J Clin 46:70, 1996.

AGE
Age has an important influence on the likelihood of being afflicted with cancer. Most carcinomas occur in
the later years of life (>55 years). Cancer is the main cause of death among women aged 40 to 79 and among
men aged 60 to 79; the decline in deaths after age 80 is due to the lower number of individuals who reach
this age. The rising incidence with age may be explained by the accumulation of somatic mutations
associated with the emergence of malignant neoplasms (discussed later). The decline in immune competence
that accompanies aging may also be a factor.
However, children are not spared; cancer accounts for slightly more than 10% of all deaths in children under
age 15 in the United States, second only to accidents. However, the types of cancers that predominate in
children are significantly different from those seen in adults. Carcinomas, the most common general
category of tumor in adults, are extraordinarily rare among children. Instead, acute leukemia and primitive
neoplasms of the central nervous system are responsible for approximately 60% of childhood cancer deaths.
The common neoplasms of infancy and childhood include the so-called small round blue cell tumors such as
neuroblastoma, Wilms tumor, retinoblastoma, acute leukemias, and rhabdomyosarcomas. These are
discussed in Chapter 10 and elsewhere in the text.
GENETIC PREDISPOSITION TO CANCER
One frequently asked question is: “My mother and father both died of cancer. Does that mean I am doomed
to get it?” Based on current knowledge, the answer must be carefully qualified.
[19,][20]
Evidence now
indicates that for a large number of cancer types, including the most common forms, there exist not only
environmental influences but also hereditary predispositions. For example, lung cancer is in most instances
clearly related to cigarette smoking, yet mortality from lung cancer has been shown to be four times greater
among nonsmoking relatives (parents and siblings) of lung cancer patients than among nonsmoking relatives
of controls (the effects of second-hand smoke may confound some of these results). Less than 10% of cancer
patients have inherited mutations that predispose to cancer, and the frequency is even lower (around 0.1%)
for certain types of tumors. Despite the low frequency, the recognition of inherited predisposition to cancer
has had a major impact on the understanding of cancer pathogenesis. Moreover, genes that are causally
associated with cancers that have a strong hereditary component are generally also involved in the much
more common sporadic forms of the same tumor. Genetic predisposition to cancer can be divided into three
categories ( Table 7-4 ).
TABLE 7-4 -- Examples of Inherited Predisposition to Cancer
INHERITED CANCER SYNDROMES (AUTOSOMAL DOMINANT)
Gene
Inherited Predisposition
RB
Retinoblastoma
p53
Li-Fraumeni syndrome (various tumors)
p16/INK4A
Melanoma
APC
Familial adenomatous polyposis/colon cancer
NF1, NF2
Neurofibromatosis 1 and 2
BRCA1, BRCA2
Breast and ovarian tumors
MEN1, RET
Multiple endocrine neoplasia 1 and 2
MSH2, MLH1, MSH6
Hereditary nonpolyposis colon cancer
PTCH
Nevoid basal cell carcinoma syndrome
PTEN
Cowden syndrome (epithelial cancers)
LKB1
Peutz-Jegher syndrome (epithelial cancers)

INHERITED CANCER SYNDROMES (AUTOSOMAL DOMINANT)
Gene
Inherited Predisposition
VHL
Renal cell carcinomas
INHERITED AUTOSOMAL RECESSIVE SYNDROMES OF DEFECTIVE DNA REPAIR
Xeroderma pigmentosum
Ataxia-telangiectasia
Bloom syndrome
Fanconi anemia
FAMILIAL CANCERS
Familial clustering of cases, but role of inherited predisposition not clear for each individual
Breast cancer
Ovarian cancer
Pancreatic cancer
Autosomal Dominant Inherited Cancer Syndromes.
Inherited cancer syndromes include several well-defined cancers in which inheritance of a single autosomal
dominant mutant gene greatly increases the risk of developing a tumor. The inherited mutation is usually a
point mutation occurring in a single allele of a tumor suppressor gene. The silencing of the second allele
occurs in somatic cells, generally as a consequence of deletion or recombination. Childhood retinoblastoma
is the most striking example in this category. Approximately 40% of retinoblastomas are inherited. Carriers
of a mutant of the RB tumor suppressor gene have a 10,000-fold increased risk of developing
retinoblastoma, usually bilateral. They also have a greatly increased risk of developing a second cancer,
particularly osteosarcoma. Familial adenomatous polyposis is an autosomal dominant hereditary disorder
caused by mutation of the adenomatous polyposis coli (APC) tumor suppressor gene. Other autosomal
dominant cancer syndromes include Li-Fraumeni syndrome resulting from germ-line mutations of the p53
gene; multiple endocrine neoplasia types 1 and 2 (MEN-1 and MEN-2) caused by mutation in the genes that
encode the menin transcription factor and the RET tyrosine kinase, respectively; hereditary nonpolyposis
colon cancer (HNPCC), a condition caused by inactivation of a DNA mismatch repair gene (also listed
below among repair defects); and several others listed in Table 7-4 .
There are several features that characterize inherited cancer syndromes:
• In each syndrome, tumors tend to arise in specific sites and tissues, although they may involve more
than one site. There is no increase in predisposition to cancers in general. For example, in MEN-2,
thyroid, parathyroid, and adrenals are involved, while in MEN-1, the pituitary, parathyroid, and
pancreas are involved. Patients with familial adenomatous polyposis develop innumerable polypoid
adenomas of the colon, and virtually 100% of those affected develop a colonic adenocarcinoma by
age 50. The one exception to this tumor specific tissue involvement is Li-Fraumeni syndrome.
• Tumors within this group are often associated with a specific marker phenotype. For example, there
may be multiple benign tumors in the affected tissue, as occurs in familial polyposis of the colon and
in MEN. Sometimes, there are abnormalities in tissue that are not the target of transformation (e.g.,
Lisch nodules and café-au-lait spots in neurofibromatosis type 1; see Chapter 27 ).
As in other autosomal dominant conditions, both incomplete penetrance and variable expressivity occur.
Defective DNA-Repair Syndromes.

Besides the dominantly inherited precancerous conditions, a group of cancer-predisposing conditions is
collectively characterized by defects in DNA repair and resultant DNA instability. These conditions
generally have an autosomal recessive pattern of inheritance. Included in this group are xeroderma
pigmentosum, ataxia-telangiectasia, and Bloom syndrome, all rare diseases characterized by genetic
instability resulting from defects in DNA-repair genes. Also included here is HNPCC, an autosomal
dominant condition caused by inactivation of a DNA mismatch repair gene.
[21]
HNPCC is the most common
cancer predisposition syndrome, increasing the susceptibility of cancer of the colon, the small intestine,
endometrium, and ovary ( Chapter 17 ).
Familial Cancers.
Besides the inherited syndromes of cancer susceptibility, cancer may occur at higher frequency in certain
families without a clearly defined pattern of transmission. Virtually all the common types of cancers that
occur sporadically have also been reported to occur in familial forms. Examples include carcinomas of
colon, breast, ovary, and brain, as well as melanomas and lymphomas. Features that characterize familial
cancers include early age at onset, tumors arising in two or more close relatives of the index case, and
sometimes, multiple or bilateral tumors. Familial cancers are not associated with specific marker
phenotypes. For example, in contrast to the familial adenomatous polyp syndrome, familial colonic cancers
do not arise in preexisting benign polyps. The transmission pattern of familial cancers is not clear. In
general, siblings have a risk between two and three times greater than unrelated individuals. Segregation
analyses of large families usually show that predisposition to the tumors is dominant, but multifactorial
inheritance cannot be easily ruled out. It is likely that familial susceptibility to cancer may depend on
multiple low-penetrance alleles, each contributing to only a small increase in the risk of tumor development.
Genome-wide association studies show great promise in identifying such alleles ( Chapter 5 ). It has been
estimated that 10% to 20% of patients with breast or ovarian cancer have a first- or second-degree relative
with one of these tumors. Although two breast cancer susceptibility genes, named BRCA1 and BRCA2, have
been identified, mutation of these genes occurs in no more than 3% of breast cancers. A similar situation
occurs in familial melanomas, in which a mutation of the p16 tumor suppressor gene has been identified.
However, mutation in this gene accounts for only about 20% of familial melanoma kindreds, suggesting that
other factors are involved in the familial predisposition.
Interactions between Genetic and Nongenetic Factors.
What can be said about the influence of heredity on the majority of malignant neoplasms? It could be argued
that they are largely of environmental origin, but lack of family history does not preclude an inherited
component. It is generally difficult to sort out the hereditary and acquired basis of a tumor, because these
factors often interact closely. The interaction between genetic and nongenetic factors is particularly complex
when tumor development depends on the action of multiple contributory genes. Even in tumors with a well-
defined inherited component, the risk of developing the tumor can be greatly influenced by nongenetic
factors. For instance, breast cancer risk in female carriers of BRCA1 or BRCA2 mutations is almost threefold
higher for women born after 1940, as compared with the risks for women born before that year.
[20]
Furthermore, the genotype can significantly influence the likelihood of developing environmentally induced
cancers. Inherited variations (polymorphisms) of enzymes that metabolize procarcinogens to their active
carcinogenic forms (see “Initiation of Carcinogenesis”) can influence the susceptibility to cancer. Of interest
in this regard are genes that encode the cytochrome P-450 enzymes. As discussed later under “Chemical
Carcinogenesis,” polymorphism at one of the P-450 loci confers inherited susceptibility to lung cancers in
cigarette smokers. More such associations are likely to be found.

NONHEREDITARY PREDISPOSING CONDITIONS
The only certain way of avoiding cancer is not to be born; to live is to incur the risk. Certain predisposing
influences, such as environment, behaviors, and clinical conditions, can increase that risk, however. For
example, regenerative, metaplastic, hyperplastic, and dysplastic proliferations are fertile soil for the origin of
a malignant tumor, because cell replication is involved in neoplastic transformation. Indeed, proliferation
may be required for neoplastic transformation in some settings, since it is proliferating cells that accumulate
the genetic lesions required for carcinogenesis.
Chronic Inflammation and Cancer.
In 1863 Virchow proposed that cancer develops at sites of chronic inflammation, and the potential
relationships between cancer and inflammation have been studied since then.
[24]
This is exemplified by the
increased risk of cancer in individuals affected by a variety of chronic inflammatory diseases of the
gastrointestinal tract ( Table 7-5 ). These include ulcerative colitis, Helicobacter pylori gastritis, viral
hepatitis, and chronic pancreatitis. Although the precise mechanisms that link inflammation and cancer
development have not been established, recent work has demonstrated that in the setting of unresolved
chronic inflammation, as occurs in viral hepatitis or chronic gastritis, the immune response may become
maladaptive, promoting tumorigenesis.
[24]
As with any cause of tissue injury, there is a compensatory
proliferation of cells so as to repair the damage. This regenerative process is aided and abetted by a plethora
of growth factors, cytokines, chemokines, and other bioactive substances produced by activated immune
cells that promote cell survival, tissue remodeling, and angiogenesis. In some cases, chronic inflammation
may increase the pool of tissue stem cells, which become subject to the effect of mutagens. These mediators
also cause genomic stress and mutations; additionally the activated immune cells produce reactive oxygen
species that are directly genotoxic. To add insult to injury, many of these mediators promote cell survival,
even in the face of genomic damage. In the short term this can be adaptive; the organism must survive, and
the damaged cells can be repaired or eliminated later. However, in chronic inflammation such behavior is
maladaptive, since it allows the creation and fixation of such mutations, eventually leading to cancer.
Whatever the precise mechanism, the link between chronic inflammation and cancer has practical
implications. For instance, expression of the enzyme cyclooxygenase-2 (COX-2), which brings about the
conversion of arachidonic acid into prostaglandins, is induced by inflammatory stimuli and is increased in
colon cancers and other tumors. The development of COX-2 inhibitors for cancer treatment is an active area
of research.
TABLE 7-5 -- Chronic Inflammatory States and Cancer
Pathologic Condition
Associated Neoplasm(s)
Etiologic Agent
Asbestosis, silicosis
Mesothelioma, lung carcinoma
Asbestos fibers, silica particles
Bronchitis
Lung carcinoma
Silica, asbestos, smoking
(nitrosamines, peroxides)
Cystitis, bladder
inflammation
Bladder carcinoma
Chronic indwelling urinary
catheters
Gingivitis, lichen planus
Oral squamous cell carcinoma
Inflammatory bowel disease Colorectal carcinoma
Lichen sclerosis
Vulvar squamous cell carcinoma
Chronic pancreatitis
Pancreatic carcinoma
Alcoholism
Hereditary pancreatitis
Pancreatic carcinoma
Mutation in trypsinogen gene
Reflux esophagitis, Barrett
esophagus
Esophageal carcinoma
Gastric acids
Sialadenitis
Salivary gland carcinoma

Pathologic Condition
Associated Neoplasm(s)
Etiologic Agent
Sjögren syndrome,
Hashimoto thyroiditis
MALT lymphoma
CANCERS ASSOCIATED WITH INFECTIOUS AGENTS
Opisthorchis, cholangitis
Cholangiosarcoma, colon carcinoma
Liver flukes (Opisthorchis
viverrini)
Bile acids
Chronic cholecystitis
Gallbladder cancer
Bacteria, gallbladder stones
Gastritis/ulcers
Gastric adenocarcinoma, MALT
Helicobacter pylori
Hepatitis
Hepatocellular carcinoma
Hepatitis B and/or C virus
Mononucleosis
B-cell non-Hodgkin lymphoma and
Hodgkin lymphoma
Epstein-Barr virus
AIDS
Non-Hodgkin lymphoma, squamous cell
carcinoma, Kaposi sarcoma
Human immunodeficiency virus,
human herpesvirus type 8
Osteomyelitis
Carcinoma in draining sinuses
Bacterial infection
Pelvic inflammatory disease,
chronic cervicitis
Ovrian carcinoma, cervical/anal
carcinoma
Gonorrhea, chlamydia, human
papillomavirus
Chronic cystitis
Bladder, liver, rectal carcinoma
Schistosomiasis
Adapted from Tlsty TD, Coussens LM: Tumor stroma and regulation of cancer development. Ann Rev
Pathol Mech Dis 1:119, 2006.
Precancerous Conditions.
Certain non-neoplastic disorders—the chronic atrophic gastritis of pernicious anemia, solar keratosis of the
skin, chronic ulcerative colitis, and leukoplakia of the oral cavity, vulva, and penis—have such a well-
defined association with cancer that they have been termed precancerous conditions. This designation is
somewhat unfortunate, because in the great majority of these lesions no malignant neoplasm emerges.
Nonetheless, the term persists because it calls attention to the increased risk. Certain forms of benign
neoplasia also constitute precancerous conditions. The villous adenoma of the colon, as it increases in size,
becomes malignant in up to 50% of cases. It might be asked: Is there not a risk with all benign neoplasms?
Although some risk may be inherent, a large cumulative experience indicates that most benign neoplasms do
not become cancerous. Nonetheless, numerous examples could be offered of cancers arising, albeit rarely, in
benign tumors—for example, a leiomyosarcoma beginning in a leiomyoma, and carcinoma appearing in
long-standing pleomorphic adenomas. Generalization is impossible, because each type of benign neoplasm
is associated with a particular level of risk ranging from virtually never to frequently. Only follow-up studies
of large series of each neoplasm can establish the level of risk, and always the question remains: Did the
cancer arise from a nonmalignant cell in the benign tumor, or did the benign tumor contain, from the outset,
a silent or indolent malignant focus?

Carcinogenic Agents and Their Cellular Interactions
More than 200 years ago the London surgeon Sir Percival Pott correctly attributed scrotal skin cancer in
chimney sweeps to chronic exposure to soot. Based on this observation, the Danish Chimney Sweeps Guild
ruled that its members must bathe daily. No public health measure since that time has achieved so much in
the control of a form of cancer. Subsequently, hundreds of chemicals have been shown to be carcinogenic in
animals. Some of the major agents are presented in Table 7-10 . A few comments are offered on a handful of
these.
TABLE 7-10 -- Major Chemical Carcinogens
DIRECT-ACTING CARCINOGENS
Alkylating Agents
β-Propiolactone
Dimethyl sulfate
Diepoxybutane
Anticancer drugs (cyclophosphamide, chlorambucil, nitrosoureas, and others)
Acylating Agents
1-Acetyl-imidazole
Dimethylcarbamyl chloride
PROCARCINOGENS THAT REQUIRE METABOLIC ACTIVATION
Polycyclic and Heterocyclic Aromatic Hydrocarbons
Benz[a]anthracene
Benzo[a]pyrene
Dibenz[a,h]anthracene
3-Methylcholanthrene
7,12-Dimethylbenz[a]anthracene
Aromatic Amines, Amides, Azo Dyes
2-Naphthylamine (β-naphthylamine)
Benzidine
2-Acetylaminofluorene
Dimethylaminoazobenzene (butter yellow)
Natural Plant and Microbial Products
Aflatoxin B
1
Griseofulvin
Cycasin
Safrole
Betel nuts
Others

Nitrosamine and amides
Vinyl chloride, nickel, chromium
Insecticides, fungicides
Polychlorinated biphenyls
Steps Involved in Chemical Carcinogenesis
As discussed earlier, carcinogenesis is a multistep process. This is most readily demonstrated in
experimental models of chemical carcinogenesis, in which the stages of initiation and progression during
cancer development were first described. The classic experiments that allowed the distinction between
initiation and promotion were performed on mouse skin and are outlined in Figure 7-41 . The following
concepts relating to the initiation-promotion sequence have emerged from these experiments:
• Initiation results from exposure of cells to a sufficient dose of a carcinogenic agent (initiator); an
initiated cell is altered, making it potentially capable of giving rise to a tumor (groups 2 and 3).
Initiation alone, however, is not sufficient for tumor formation (group 1).
• Initiation causes permanent DNA damage (mutations). It is therefore rapid and irreversible and has
“memory.” This is illustrated by group 3, in which tumors were produced even if the application of
the promoting agent was delayed for several months after a single application of the initiator.
• Promoters can induce tumors in initiated cells, but they are nontumorigenic by themselves (group 5).
Furthermore, tumors do not result when the promoting agent is applied before, rather than after, the
initiating agent (group 4). This indicates that, in contrast to the effects of initiators, the cellular
changes resulting from the application of promoters do not affect DNA directly and are reversible.
As discussed later, promoters enhance the proliferation of initiated cells, an effect that may contribute
to the development of additional mutations in these cells. That the effects of promoters are reversible
is further documented in group 6, in which tumors failed to develop in initiated cells if the time
between multiple applications of the promoter was sufficiently extended.
Although the concepts of initiation and promotion have been derived largely from experiments involving
induction of skin cancer in mice, these stages are also discernible in the development of cancers of the liver,
urinary bladder, breast, colon, and respiratory tract. With this brief overview of two major steps in
carcinogenesis, we can examine initiation and promotion in more detail ( Fig. 7-42 ). All initiating chemical
carcinogens are highly reactive electrophiles (have electron-deficient atoms) that can react with nucleophilic
(electron-rich) sites in the cell. Their targets are DNA, RNA, and proteins, and in some cases these
interactions cause cell death. Initiation, obviously, inflicts nonlethal damage on the DNA that cannot be
repaired. The mutated cell then passes on the DNA lesions to its daughter cells. Chemicals that can cause
initiation of carcinogenesis can be classified into two categories: direct acting and indirect acting.
Direct-Acting Agents
Direct-acting agents require no metabolic conversion to become carcinogenic. Most of them are weak
carcinogens but are important because some are cancer chemotherapeutic drugs (e.g., alkylating agents) that
have successfully cured, controlled, or delayed recurrence of certain types of cancer (e.g., leukemia,

lymphoma, and ovarian carcinoma), only to evoke later a second form of cancer, usually acute myeloid
leukemia. The risk of induced cancer is low, but its existence dictates judicious use of such agents.
Indirect-Acting Agents
The designation indirect-acting agent refers to chemicals that require metabolic conversion to an ultimate
carcinogen before they become active. Some of the most potent indirect chemical carcinogens—the
polycyclic hydrocarbons—are present in fossil fuels. Others, for example, benzo[a]pyrene and other
carcinogens, are formed in the high-temperature combustion of tobacco in cigarette smoking. These
products are implicated in the causation of lung cancer in cigarette smokers. Polycyclic hydrocarbons may
also be produced from animal fats during the process of broiling meats and are present in smoked meats and
fish. The principal active products in many hydrocarbons are epoxides, which form covalent adducts
(addition products) with molecules in the cell, principally DNA, but also with RNA and proteins.
The aromatic amines and azo dyes are another class of indirect-acting carcinogens that were widely used in
the past in the aniline dye and rubber industrie
Most chemical carcinogens require metabolic activation for conversion into ultimate carcinogens. Other
metabolic pathways may lead to the inactivation (detoxification) of the procarcinogen or its derivatives.
Thus, the carcinogenic potency of a chemical is determined not only by the inherent reactivity of its
electrophilic derivative but also by the balance between metabolic activation and inactivation reactions.
Most of the known carcinogens are metabolized by cytochrome P-450–dependent mono-oxygenases. The
genes that encode these enzymes are quite polymorphic, and the activity and inducibility of these enzymes
have been shown to vary among different individuals. Because these enzymes are essential for the activation
of procarcinogens, the susceptibility to carcinogenesis is regulated in part by polymorphisms in the genes
that encode these enzymes. Thus, it may be possible to assess cancer risk in a given individual by genetic
analysis of such enzyme polymorphisms.
[147]
The metabolism of polycyclic aromatic hydrocarbons, such as benzo[a]pyrene by the product of the P-450
gene, CYP1A1, provides an instructive example. Approximately 10% of the white population has a highly
inducible form of this enzyme that is associated with an increased risk of lung cancer in smokers.
Light
smokers with the susceptible genotype CYP1A1 have a sevenfold higher risk of developing lung cancer,
compared with smokers without the permissive genotype. Not all variations in the activation or
detoxification of carcinogens are genetically determined. Age, sex, and nutritional status also determine the
internal dose of toxicants produced and hence influence the risk of cancer development in a particular
individual.
RADIATION CARCINOGENESIS
Radiant energy, whether in the form of the UV rays of sunlight or as ionizing electromagnetic and
particulate radiation, is a well-established carcinogen. UV light is clearly implicated in the causation of skin
cancers, and ionizing radiation exposure from medical or occupational exposure, nuclear plant accidents,
and atomic bomb detonations has produced a variety of cancers. Although the contribution of radiation to
the total human burden of cancer is probably small, the well-known latency of damage caused by radiant
energy and its cumulative effect require extremely long periods of observation and make it difficult to
ascertain its full significance. An increased incidence of breast cancer has become apparent decades later
among women exposed during childhood to atomic bomb tests. The incidence peaked during 1988–1992
and then declined. Moreover, radiation's possible additive or synergistic effects with other potential
carcinogenic influences add another dimension to the picture.
Ultraviolet Rays
There is ample evidence from epidemiologic studies that UV rays derived from the sun cause an increased
incidence of squamous cell carcinoma, basal cell carcinoma, and possibly melanoma of the skin.
[153]
The

degree of risk depends on the type of UV rays, the intensity of exposure, and the quantity of the light-
absorbing “protective mantle” of melanin in the skin. Persons of European origin who have fair skin that
repeatedly becomes sunburned but stalwartly refuses to tan and who live in locales receiving a great deal of
sunlight (e.g., Queensland, Australia, close to the equator) have among the highest incidence of skin cancers
(melanomas, squamous cell carcinomas, and basal cell carcinomas) in the world. Nonmelanoma skin cancers
are associated with total cumulative exposure to UV radiation, whereas melanomas are associated with
intense intermittent exposure—as occurs with sunbathing. The UV portion of the solar spectrum can be
divided into three wavelength ranges: UVA (320–400 nm), UVB (280–320 nm), and UVC (200–280 nm).
Of these, UVB is believed to be responsible for the induction of cutaneous cancers. UVC, although a potent
mutagen, is not considered significant because it is filtered out by the ozone shield around the earth (hence
the concern about ozone depletion).
The carcinogenicity of UVB light is attributed to its formation of pyrimidine dimers in DNA. This type of
DNA damage is repaired by the nucleotide excision repair pathway. There are five steps in nucleotide
excision repair, and in mammalian cells the process may involve 30 or more proteins. It is postulated that
with excessive sun exposure, the capacity of the nucleotide excision repair pathway is overwhelmed, and
error-prone nontemplated DNA-repair mechanisms become operative that provide for the survival of the cell
at the cost of genomic mutations that in some instances, lead to cancer. The importance of the nucleotide
excision repair pathway of DNA repair is most graphically illustrated by the high frequency of cancers in
individuals with the hereditary disorder xeroderma pigmentosum (discussed previously)
Ionizing Radiation
Electromagnetic (x-rays, γ rays) and particulate (α particles, β particles, protons, neutrons) radiations are all
carcinogenic. The evidence is so voluminous that a few examples suffice. Many individuals pioneering the
use of x-rays developed skin cancers. Miners of radioactive elements in central Europe and the Rocky
Mountain region of the United States have a tenfold increased incidence of lung cancers compared to the
rest of the population. Most telling is the follow-up of survivors of the atomic bombs dropped on Hiroshima
and Nagasaki. Initially there was a marked increase in the incidence of leukemias—principally acute and
chronic myelogenous leukemia—after an average latent period of about 7 years. Subsequently the incidence
of many solid tumors with longer latent periods (e.g., breast, colon, thyroid, and lung) increased.
MICROBIAL CARCINOGENESIS
Many RNA and DNA viruses have proved to be oncogenic in animals as disparate as frogs and primates.
Despite intense scrutiny, however, only a few viruses have been linked with human cancer. Our discussion
focuses on human oncogenic viruses as well as the emerging role of the bacterium Helicobacter pylori in
gastric cancer.
Oncogenic RNA Viruses
Human T-Cell Leukemia Virus Type 1.
Although the study of animal retroviruses has provided spectacular insights into the molecular basis of
cancer, only one human retrovirus, human T-cell leukemia virus type 1 (HTLV-1), is firmly implicated in
the causation of cancer in humans.
HTLV-1 causes a form of T-cell leukemia/lymphoma that is endemic in certain parts of Japan and the
Caribbean basin but is found sporadically elsewhere, including the United States.
[155]
Similar to the human
immunodeficiency virus, which causes acquired immunodeficiency syndrome (AIDS), HTLV-1 has tropism
for CD4+ T cells, and hence this subset of T cells is the major target for neoplastic transformation. Human
infection requires transmission of infected T cells via sexual intercourse, blood products, or breastfeeding.
Leukemia develops in only 3% to 5% of the infected individuals after a long latent period of 40 to 60 years.
There is little doubt that HTLV-1 infection of T lymphocytes is necessary for leukemogenesis, but the
molecular mechanisms of transformation are not entirely clear. In contrast to several murine retroviruses,

HTLV-1 does not contain an oncogene, and no consistent integration next to a proto-oncogene has been
discovered. In leukemic cells, however, viral integration shows a clonal pattern. In other words, although the
site of viral integration in host chromosomes is random (the viral DNA is found at different locations in
different cancers), the site of integration is identical within all cells of a given cancer. This would not occur
if HTLV-1 were merely a passenger that infects cells after transformation. The HTLV-1 genome contains
the gag, pol, env, and long-terminal-repeat regions typical of other retroviruses, but, in contrast to other
leukemia viruses, it contains another region, referred to as tax. It seems that the secrets of its transforming
activity are locked in the tax gene.
[156]
The product of this gene is essential for viral replication, because it
stimulates transcription of viral mRNA by acting on the 5′ long terminal repeat. It is now established that the
Tax protein can also activate the transcription of several host cell genes involved in proliferation and
differentiation of T cells. These include the immediate early gene FOS, genes encoding interleukin-2 (IL-2)
and its receptor, and the gene for the myeloid growth factor granulocyte-macrophage colony-stimulating
factor. In addition, Tax protein inactivates the cell cycle inhibitor p16/INK4a and enhances cyclin D
activation, thus dysregulating the cell cycle. Tax also activates NF-κb, a transcription factor that regulates a
host of genes, including pro-survival/anti-apoptotic genes. Another mechanism by which Tax may
contribute to malignant transformation is through genomic instability. Recent data show that Tax interferes
with DNA-repair functions and inhibits ATM-mediated cell cycle checkpoints activated by DNA damage.
Oncogenic DNA Viruses
As with RNA viruses, several oncogenic DNA viruses that cause tumors in animals have been identified. Of
the various human DNA viruses, four—HPV, Epstein-Barr virus (EBV), hepatitis B virus (HBV), and
Kaposi sarcoma herpesvirus, also called human herpesvirus 8—have been implicated in the causation of
human cancer. A fifth virus, Merkel cell polyomavirus, has been identified in Merkel cell carcinomas and
may soon join the rogue's gallery; it is described in Chapter 25 . Kaposi sarcoma herpesvirus is discussed in
Chapters 6 and 11 . Though not a DNA virus, HCV is also associated with cancer and is discussed briefly
here.
Human Papillomavirus.
At least 70 genetically distinct types of HPV have been identified. Some types (e.g., 1, 2, 4, and 7) cause
benign squamous papillomas (warts) in humans ( Chapters 19 and 22 . By contrast, high-risk HPVs (e.g.,
types 16 and 18) have been implicated in the genesis of several cancers, particularly squamous cell
carcinoma of the cervix and anogenital region. Thus, cervical cancer is a sexually transmitted disease,
caused by transmission of HPV. In addition, at least 20% of oropharyngeal cancers are associated with HPV.
In contrast to cervical cancers, genital warts have low malignant potential and are associated with low-risk
HPVs, predominantly HPV-6 and HPV-11. Interestingly, in benign warts the HPV genome is maintained in
a nonintegrated episomal form, while in cancers the HPV genome is integrated into the host genome,
suggesting that integration of viral DNA is important for malignant transformation. As with HTLV-1, the
site of viral integration in host chromosomes is random, but the pattern of integration is clonal. Cells in
which the viral genome has integrated show significantly more genomic instability. Furthermore, since the
integration site is random there is no consistent association with a host proto-oncogene. Rather, integration
interrupts the viral DNA within the E1/E2 open reading frame, leading to loss of the E2 viral repressor and
overexpression of the oncoproteins E6 and E7.
To summarize, high-risk HPV types express oncogenic proteins that inactivate tumor suppressors, activate
cyclins, inhibit apoptosis, and combat cellular senescence. Thus, it is evident that many of the hallmarks of
cancer discussed earlier are driven by HPV proteins. The primacy of HPV infection in the causation of
cervical cancer is confirmed by the effectiveness of anti-HPV vaccines in preventing cervical cancer.
However, infection with HPV itself is not sufficient for carcinogenesis. For example, when human
keratinocytes are transfected with DNA from HPV types 16, 18, or 31 in vitro, they are immortalized but do
not form tumors in experimental animals. Co-transfection with a mutated RAS gene results in full malignant
transformation. In addition to such genetic co-factors, HPV in all likelihood also acts in concert with
environmental factors. These include cigarette smoking, coexisting microbial infections, dietary

deficiencies, and hormonal changes, all of which have been implicated in the pathogenesis of cervical
cancers. A high proportion of women infected with HPV clear the infection by immunological mechanisms,
but some do not for unknown reasons.
Epstein-Barr Virus.
EBV, a member of the herpes family, has been implicated in the pathogenesis of several human tumors: the
African form of Burkitt lymphoma; B-cell lymphomas in immunosuppressed individuals (particularly in
those with HIV infection or undergoing immunosuppressive therapy after organ transplantation); a subset of
Hodgkin lymphoma; nasopharyngeal and some gastric carcinomas and rare forms of T cell lymphomas and
natural killer (NK) cell lymphomas. Except for nasopharyngeal carcinoma, all others are B-cell tumors.
Burkitt lymphoma is a neoplasm of B lymphocytes that is the most common childhood tumor in central
Africa and New Guinea. A morphologically identical lymphoma occurs sporadically throughout the world.
The association between endemic Burkitt lymphoma and EBV is quite strong.
• More than 90% of African tumors carry the EBV genome.
• One hundred percent of the patients have elevated antibody titers against viral capsid antigens.
• Serum antibody titers against viral capsid antigens are correlated with the risk of developing the
tumor.
Although EBV is intimately involved in the causation of Burkitt lymphoma, several observations suggest
that additional factors must also be involved. (1) EBV infection is not limited to the geographic locales
where Burkitt lymphoma is found, but it is a ubiquitous virus that asymptomatically infects almost all
humans worldwide. (2) The EBV genome is found in only 15% to 20% of sufferers of Burkitt lymphoma
outside Africa. (3) There are significant differences in the patterns of viral gene expression in EBV-
transformed (but not tumorigenic) B-cell lines and Burkitt lymphoma cells. Most notably, Burkitt lymphoma
cells do not express LMP-1, EBNA2, and other EBV proteins that drive B-cell growth and immortalization.
In summary, in the case of Burkitt lymphoma, it seems that EBV is not directly oncogenic, but by acting as a
polyclonal B-cell mitogen, it sets the stage for the acquisition of the t(8;14) translocation and other
mutations, which ultimately release the cells from normal growth regulation. In normal individuals, EBV
infection is readily controlled by effective immune responses directed against viral antigens expressed on the
cell membranes. Hence, the vast majority of infected individuals remain asymptomatic or develop self-
limited infectious mononucleosis. In regions of Africa where Burkitt lymphoma is endemic, poorly
understood cofactors (e.g., chronic malaria) may favor the acquisition of genetic events (e.g., the t(8;14)
translocation) that lead to transformation.
The role played by EBV is more direct in B-cell lymphomas in immunosuppressed patients. Some persons
with AIDS and those who receive long-term immunosuppressive therapy for preventing allograft rejection
present with multifocal B-cell tumors within lymphoid tissue or in the central nervous system. These tumors
are polyclonal at the outset but can develop into monoclonal neoplasms. In contrast to Burkitt lymphoma,
the tumors in immunosuppressed patients uniformly express LMP-1 and EBNA2, that are recognized by
cytotoxic T cells. These potentially lethal proliferations can be subdued if the immunological status of the
host improves, as may occur with withdrawal of immunosuppressive drugs in transplant recipients.
Nasopharyngeal carcinoma is also associated with EBV infection. This tumor is endemic in southern China,
in some parts of Africa, and in the Inuit population of the Arctic. In contrast to Burkitt lymphoma, 100% of
nasopharyngeal carcinomas obtained from all parts of the world contain EBV DNA.The viral integration in
the host cells is clonal, thus ruling out the possibility that EBV infection occurred after tumor development.
Antibody titers to viral capsid antigens are greatly elevated, and in endemic areas patients develop IgA

antibodies before the appearance of the tumor. The 100% correlation between EBV and nasopharyngeal
carcinoma suggests that EBV plays a role in the genesis of this tumor, but (as with Burkitt tumor) the
restricted geographic distribution indicates that genetic or environmental cofactors, or both, also contribute
to tumor development. LMP-1 is expressed in epithelial cells as well. In these cells, as in B cells, LMP-1
activates the NF-κB pathway. Furthermore, LMP-1 induces the expression of pro-angiogenic factors such as
VEGF, FGF-2, MMP9, and COX2, which may contribute to oncogenesis. The relationship of EBV to the
pathogenesis of Hodgkin lymphoma is discussed in Chapter 13 .
Hepatitis B and C Viruses.
Epidemiologic studies strongly suggest a close association between HBV infection and the occurrence of
liver cancer . It is estimated that 70% to 85% of hepatocellular carcinomas worldwide are due to infection
with HBV or HCV.
HBV is endemic in countries of the Far East and Africa; correspondingly, these areas
have the highest incidence of hepatocellular carcinoma. Despite compelling epidemiologic and experimental
evidence, the mode of action of these viruses in liver tumorigenesis is not fully elucidated. The HBV and
HCV genomes do not encode any viral oncoproteins, and although the HBV DNA is integrated within the
human genome, there is no consistent pattern of integration in liver cells. Indeed, the oncogenic effects of
HBV and HCV are multifactorial, but the dominant effect seems to be immunologically mediated chronic
inflammation with hepatocyte death leading to regeneration and genomic damage. Although the immune
system is generally thought to be protective, recent work has demonstrated that in the setting of unresolved
chronic inflammation, as occurs in viral hepatitis or chronic gastritis caused by H. pylori (see below), the
immune response may become maladaptive, promoting tumorigenesis.
As with any cause of hepatocellular injury, chronic viral infection leads to the compensatory proliferation of
hepatocytes. This regenerative process is aided and abetted by a plethora of growth factors, cytokines,
chemokines, and other bioactive substances that are produced by activated immune cells and promote cell
survival, tissue remodeling, and angiogenesis ( Chapter 3 ). The activated immune cells also produce other
mediators, such as reactive oxygen species, that are genotoxic and mutagenic. One key molecular step seems
to be activation of the NF-κB pathway in hepatocytes in response to mediators derived from the activated
immune cells. Activation of the NF-κB pathway within hepatocytes blocks apoptosis, allowing the dividing
hepatocytes to incur genotoxic stress and to accumulate mutations. Although this seems to be the dominant
mechanism in the pathogenesis of viral-induced hepatocellular carcinoma, both HBV and HCV also contain
proteins within their genomes that may more directly promote the development of cancer. The HBV genome
contains a gene known as HBx that can directly or indirectly activate a variety of transcription factors and
several signal transduction pathways. In addition, viral integration can cause secondary rearrangements of
chromosomes, including multiple deletions that may harbor unknown tumor suppressor genes.
Though not a DNA virus, HCV is also strongly linked to the pathogenesis of liver cancer. The molecular
mechanisms used by HCV are less well defined than are those of HBV. In addition to chronic liver cell
injury and compensatory regeneration, components of the HCV genome, such as the HCV core protein, may
have a direct effect on tumorigenesis, possibly by activating a variety of growth-promoting signal
transduction pathways.
Helicobacter pylori
First incriminated as a cause of peptic ulcers, H. pylori now has acquired the dubious distinction of being the
first bacterium classified as a carcinogen. Indeed, H. pylori infection is implicated in the genesis of both
gastric adenocarcinomas and gastric lymphomas.
H. pylori is associated with an increased risk for the development of gastric lymphomas as well. The gastric
lymphomas are of B-cell origin, and because the tumors recapitulate some of the features of normal Peyer's
patches, they are often called lymphomas of mucosa-associated lymphoid tissue, or MALTomas (also
discussed in Chapters 13 and 17 . Their molecular pathogenesis is incompletely understood but seems to
involve strain-specific H. pylori factors, as well as host genetic factors, such as polymorphisms in the
promoters of inflammatory cytokines such as IL-1β and tumor necrosis factor (TNF). It is thought that H.

pylori infection leads to the appearance of H. pylori–reactive T cells, which in turn stimulate a polyclonal B-
cell proliferation. In chronic infections, currently unknown mutations may be acquired that give individual
cells a growth advantage. These cells grow out into a monoclonal “MALToma” that nevertheless remains
dependent on T-cell stimulation of B-cell pathways that activate the transcription factor NF-κB. At this
stage, eradication of H. pylori by antibiotic therapy “cures” the lymphoma by removing the antigenic
stimulus for T cells. At later stages, however, additional mutations may be acquired, such as an (11;18)
translocation, that cause NF-κB to be activated constitutively. At this point, the MALToma no longer
requires the antigenic stimulus of the bacterium for growth and survival and develops the capacity to spread
beyond the stomach to other tissues.
Host Defense against Tumors—Tumor Immunity
The idea that tumors are not entirely self and may be recognized by the immune system was conceived by
Paul Ehrlich, who proposed that immune recognition of autologous tumor cells may be capable of
eliminating tumors. Subsequently, Lewis Thomas and Macfarlane Burnet formalized this concept by coining
the term immune surveillance, which implies that a normal function of the immune system is to survey the
body for emerging malignant cells and destroy them. This idea has been supported by many observations—
the occurrence of lymphocytic infiltrates around tumors and in lymph nodes draining sites of cancer;
experimental results, mostly with transplanted tumors; the increased incidence of some cancers in
immunodeficient individuals; and the direct demonstration of tumor-specific T cells and antibodies in
patients. The fact that cancers occur in immunocompetent individuals suggests that immune surveillance is
imperfect; however, that some tumors escape such policing does not preclude the possibility that others may
have been aborted. The concept of tumor immune surveillance has recently been expanded to encompass not
only the protective role of the immune system in tumor development but also the effect of the immune
system in selecting for tumor variants. These variants have reduced immunogenicity and can more easily
escape immunological detection and rejection. The term cancer immunoediting is now being used to
describe the effects of the immune system
TUMOR ANTIGENS
Antigens that elicit an immune response have been demonstrated in many experimentally induced tumors
and in some human cancers.Initially, they were broadly classified into two categories based on their patterns
of expression: tumor-specific antigens, which are present only on tumor cells and not on any normal cells,
and tumor-associated antigens, which are present on tumor cells and also on some normal cells. This
classification, however, is imperfect because many antigens thought to be tumor-specific turned out to be
expressed by some normal cells as well. The modern classification of tumor antigens is based on their
molecular structure and source.
The early attempts to purify and characterize tumor antigens relied on producing monoclonal antibodies
specific for tumor cells and defining the antigens that these antibodies recognized. An important advance in
the field was the development of techniques for identifying tumor antigens that were recognized by
cytotoxic T lymphocytes (CTLs), because CTLs are the major immune defense mechanism against tumors.
Recall that CTLs recognize peptides derived from cytoplasmic proteins that are displayed bound to class I
major histocompatibility complex (MHC) molecules ( Chapter 6 ). Below we describe the main classes of
tumor antigens.
Overexpressed or Aberrantly Expressed Cellular Proteins.
Tumor antigens may be normal cellular proteins that are abnormally expressed in tumor cells and elicit
immune responses. In a subset of human melanomas some tumor antigens are structurally normal proteins
that are produced at low levels in normal cells and overexpressed in tumor cells. One such antigen is
tyrosinase, an enzyme involved in melanin biosynthesis that is expressed only in normal melanocytes and
melanomas.
[179]
T cells from melanoma patients recognize peptides derived from tyrosinase, raising the

possibility that tyrosinase vaccines may stimulate such responses to melanomas; clinical trials with these
vaccines are ongoing. It may be surprising that these patients are able to respond to a normal self-antigen.
The probable explanation is that tyrosinase is normally produced in such small amounts and in so few cells
that it is not recognized by the immune system and fails to induce tolerance.
Another group, the “cancer-testis” antigens, are encoded by genes that are silent in all adult tissues except
the testis—hence their name. Although the protein is present in the testis it is not expressed on the cell
surface in an antigenic form, because sperm do not express MHC class I antigens. Thus, for all practical
purposes these antigens are tumor specific. Prototypic of this group is the melanoma antigen gene (MAGE)
family. Although originally described in melanomas, MAGE antigens are expressed by a variety of tumor
types. For example, MAGE-1 is expressed on 37% of melanomas and a variable number of lung, liver,
stomach, and esophageal carcinomas.
[180]
Similar antigens called GAGE, BAGE, and RAGE have been
detected in other tumors.
Tumor Antigens Produced by Oncogenic Viruses.
As we have discussed, several viruses are associated with cancers. Not surprisingly, these viruses produce
proteins that are recognized as foreign by the immune system. The most potent of these antigens are proteins
produced by latent DNA viruses; examples in humans include HPV and EBV. There is abundant evidence
that CTLs recognize antigens of these viruses and that a competent immune system plays a role in
surveillance against virus-induced tumors because of its ability to recognize and kill virus-infected cells. In
fact, the concept of immune surveillance against tumors is best established for DNA virus–induced tumors.
Indeed, vaccines against HPV antigens are effective in preventing cervical cancers in young females.
Oncofetal Antigens.
Oncofetal antigens are proteins that are expressed at high levels on cancer cells and in normal developing
(fetal) but not adult tissues. It is believed that the genes encoding these proteins are silenced during
development and are derepressed upon malignant transformation. Oncofetal antigens were identified with
antibodies raised in other species, and their main importance is that they provide markers that aid in tumor
diagnosis. As techniques for detecting these antigens have improved, it has become clear that their
expression in adults is not limited to tumors. Amounts of these proteins are increased in tissues and in the
circulation in various inflammatory conditions, and they are found in small quantities even in normal tissues.
There is no evidence that oncofetal antigens are important inducers or targets of antitumor immunity. The
two most thoroughly characterized oncofetal antigens are carcinoembryonic antigen (CEA) and α-
fetoprotein (AFP). These are discussed in the section on “Tumor Markers”.
ANTITUMOR EFFECTOR MECHANISMS
Cell-mediated immunity is the dominant antitumor mechanism in vivo. Although antibodies can be made
against tumors, there is no evidence that they play a protective role under physiologic conditions. The
cellular effectors that mediate immunity were described in Chapter 6 , so it is necessary here only to
characterize them briefly.
• Cytotoxic T lymphocytes: The antitumor effect of cytotoxic T cells reacting against tumor antigens is
well established in experimentally induced tumors. In humans, CD8+ CTLs play a protective role
against virus-associated neoplasms (e.g., EBV- and HPV-induced tumors) and have been
demonstrated in the blood and tumor infiltrates of cancer patients. In some cases, such CD8+ T cells
do not develop spontaneously in vivo but can be generated by immunization with tumor antigen–
pulsed dendritic cells.
• Natural killer cells: NK cells are lymphocytes that are capable of destroying tumor cells without
prior sensitization and thus may provide the first line of defense against tumor cells.
[181]
After
activation with IL-2 and IL-15, NK cells can lyse a wide range of human tumors, including many that
seem to be nonimmunogenic for T cells. T cells and NK cells seem to provide complementary

antitumor mechanisms. Tumors that fail to express MHC class I antigens cannot be recognized by T
cells, but these tumors may trigger NK cells because the latter are inhibited by recognition of normal
autologous class I molecules ( Chapter 6 ). The triggering receptors on NK cells are extremely
diverse and belong to several gene families. NKG2D proteins expressed on NK cells and some T
cells are important activating receptors. They recognize stress-induced antigens that are expressed on
tumor cells and cells that have incurred DNA damage and are at risk for neoplastic transformation.
• Macrophages: Activated macrophages exhibit cytotoxicity against tumor cells in vitro. T cells, NK
cells, and macrophages may collaborate in antitumor reactivity, because interferon-γ, a cytokine
secreted by T cells and NK cells, is a potent activator of macrophages. Activated macrophages may
kill tumors by mechanisms similar to those used to kill microbes (e.g., production of reactive oxygen
metabolites; Chapter 2 ) or by secretion of TNF.
• Antibodies: Although there is no evidence for the protective effects of antitumor antibodies against
spontaneous tumors, administration of monoclonal antibodies against tumor cells can be
therapeutically effective. A monoclonal antibody against CD20, a B-cell surface antigen, is widely
used for treatment of lymphomas.
Clinical Aspects of Neoplasia
Ultimately the importance of neoplasms lies in their effects on patients. Although malignant tumors are of
course more threatening than benign tumors, any tumor, even a benign one, may cause morbidity and
mortality. Indeed, both malignant and benign tumors may cause problems because of (1) location and
impingement on adjacent structures, (2) functional activity such as hormone synthesis or the development of
paraneoplastic syndromes, (3) bleeding and infections when the tumor ulcerates through adjacent surfaces,
(4) symptoms that result from rupture or infarction, and (5) cachexia or wasting.
Local and Hormonal Effects
Location is crucial in both benign and malignant tumors. A small (1-cm) pituitary adenoma, though benign
and possibly nonfunctional, can compress and destroy the surrounding normal gland and thus lead to serious
hypopituatarism. Cancers arising within or metastatic to an endocrine gland may cause an endocrine
insufficiency by destroying the gland. Neoplasms in the gut, both benign and malignant, may cause
obstruction as they enlarge. Infrequently, peristaltic movement telescopes the neoplasm and its affected
segment into the downstream segment, producing an obstructing intussusception ( Chapter 17 ).
Hormone production is seen with benign and malignant neoplasms arising in endocrine glands. Such
functional activity is more typical of benign than of malignant tumors, which may be sufficiently
undifferentiated to have lost such capability. A benign beta-cell adenoma of the pancreatic islets less than 1
cm in diameter may produce sufficient insulin to cause fatal hypoglycemia. In addition, nonendocrine
tumors may elaborate hormones or hormone-like products and give rise to paraneoplastic syndromes
(discussed later). The erosive and destructive growth of cancers or the expansile pressure of a benign tumor
on any natural surface, such as the skin or mucosa of the gut, may cause ulcerations, secondary infections,
and bleeding. Melena (blood in the stool) and hematuria, for example, are characteristic of neoplasms of the
gut and urinary tract. Neoplasms, benign as well as malignant, may cause problems in varied ways, but all
are far less common than the cachexia of malignancy.
Cancer Cachexia
Individuals with cancer commonly suffer progressive loss of body fat and lean body mass accompanied by
profound weakness, anorexia, and anemia, referred to as cachexia. Unlike starvation, the weight loss seen in
cachexia results equally from loss of fat and lean muscle. There is some correlation between the tumor
burden and the severity of the cachexia. However, cachexia is not caused by the nutritional demands of the
tumor. In persons with cancer, the basal metabolic rate is increased, despite reduced food intake. This is in
contrast to the lower metabolic rate that occurs as an adaptational response in starvation. Although patients

with cancer are often anorexic, cachexia probably results from the action of soluble factors such as cytokines
produced by the tumor and the host rather than reduced food intake. The basis of these metabolic
abnormalities is not fully understood. It is suspected that TNF produced by macrophages in response to
tumor cells or by the tumor cells themselves mediates cachexia. TNF at high concentrations may mobilize
fats from tissue stores and suppress appetite; both activities would contribute to cachexia. Other cytokines,
such as IL-1, interferon-γ, and leukemia inhibitory factor, synergize with TNF. Additionally, other soluble
factors produced by tumors, such as proteolysis-inducing factor and a lipid-mobilizing factor, increase the
catabolism of muscle and adipose tissue. These factors reduce protein synthesis by decreasing m-RNA
translation and by stimulating protein catabolism through the activation of the ATP-dependent ubiquitin-
proteasome pathway. It is now thought that there is a balance between factors that regulate muscle
hypertrophy, such as IGF, and factors that regulate muscle catabolism. In cachexia these homeostatic
mechanisms are disrupted, tilting the scales toward cachectic factors. There is currently no satisfactory
treatment for cancer cachexia other than removal of the underlying cause, the tumor. However, cachexia
clearly hampers effective chemotherapy, by reducing the dosages that can be given. Furthermore, it has been
estimated that a third of deaths of cancer are attributable to cachexia, rather than directly due to the tumor
burden itself. Identification of the molecular mechanisms involved in cancer cachexia may allow treatment
of cachexia itself.
Paraneoplastic Syndromes
Symptom complexes in cancer-bearing individuals that cannot readily be explained, either by the local or
distant spread of the tumor or by the elaboration of hormones indigenous to the tissue from which the tumor
arose, are known as paraneoplastic syndromes.These occur in about 10% of persons with malignant disease.
Despite their relative infrequency, paraneoplastic syndromes are important to recognize, for several reasons:
• They may represent the earliest manifestation of an occult neoplasm.
• In affected patients they may represent significant clinical problems and may even be lethal.
• They may mimic metastatic disease and therefore confound treatment.
A classification of paraneoplastic syndromes and their presumed origins is presented in Table 7-11 . A few
comments on some of the more common and interesting syndromes follow.
TABLE 7-11 -- Paraneoplastic Syndromes
Clinical Syndromes
Major Forms of Underlying
Cancer
Causal Mechanism
ENDOCRINOPATHIES
Cushing syndrome
Small-cell carcinoma of lung
ACTH or ACTH-like substance
Pancreatic carcinoma
Neural tumors
Syndrome of inappropriate
antidiuretic hormone secretion
Small-cell carcinoma of lung;
intracranial neoplasms
Antidiuretic hormone or atrial
natriuretic hormones
Hypercalcemia
Squamous cell carcinoma of lung Parathyroid hormone–related protein
(PTHRP), TGF-α, TNF, IL-1
Breast carcinoma
Renal carcinoma
Adult T-cell leukemia/lymphoma
Hypoglycemia
Ovarian carcinoma
Fibrosarcoma
Insulin or insulin-like substance

Clinical Syndromes
Major Forms of Underlying
Cancer
Causal Mechanism
Other mesenchymal sarcomas
Carcinoid syndrome
Hepatocellular carcinoma
Bronchial adenoma (carcinoid)
Serotonin, bradykinin
Pancreatic carcinoma
Polycythemia
Gastric carcinoma
Renal carcinoma
Erythropoietin
Cerebellar hemangioma
Hepatocellular carcinoma
NERVE AND MUSCLE SYNDROMES
Myasthenia
Bronchogenic carcinoma
Immunological
Disorders of the central and
peripheral nervous system
Breast carcinoma
DERMATOLOGIC DISORDERS
Acanthosis nigricans
Gastric carcinoma
Immunological; secretion of
epidermal growth factor
Lung carcinoma
Uterine carcinoma
Dermatomyositis
Bronchogenic, breast carcinoma Immunological
OSSEOUS, ARTICULAR, AND SOFT-TISSUE CHANGES
Hypertrophic osteoarthropathy
and clubbing of the fingers
Bronchogenic carcinoma
Unknown
VASCULAR AND HEMATOLOGIC CHANGES
Venous thrombosis (Trousseau
phenomenon)
Pancreatic carcinoma
Tumor products (mucins that activate
clotting)
Bronchogenic carcinoma
Other cancers
Nonbacterial thrombotic
endocarditis
Advanced cancers
Hypercoagulability
Red cell aplasia
Thymic neoplasms
Unknown
OTHERS
Nephrotic syndrome
Various cancers
Tumor antigens, immune complexes
ACTH, adrenocorticotropic hormone; IL, interleukin; TGF, transforming growth factor; TNF, tumor
necrosis factor.
The endocrinopathies are frequently encountered paraneoplastic syndromes. Because the cancer cells are not
of endocrine origin, the functional activity is referred to as ectopic hormone production. Cushing syndrome
is the most common endocrinopathy. Approximately 50% of individuals with this endocrinopathy have
carcinoma of the lung, chiefly the small-cell type. It is caused by excessive production of corticotropin or
corticotropin-like peptides. The precursor of corticotropin is a large molecule known as pro-
opiomelanocortin. Lung cancer patients with Cushing syndrome have elevated serum levels of pro-

opiomelanocortin and of corticotropin. The former is not found in serum of patients with excess
corticotropin produced by the pituitary.
Hypercalcemia is probably the most common paraneoplastic syndrome; overtly symptomatic hypercalcemia
is most often related to some form of cancer rather than to hyperparathyroidism. Two general processes are
involved in cancer-associated hypercalcemia: (1) osteolysis induced by cancer, whether primary in bone,
such as multiple myeloma, or metastatic to bone from any primary lesion, and (2) the production of calcemic
humoral substances by extraosseous neoplasms. Hypercalcemia due to skeletal metastases is not a
paraneoplastic syndrome.
Several humoral factors have been associated with paraneoplastic hypercalcemia of malignancy. The most
important, parathyroid hormone–related protein (PTHRP), is a molecule related to, but distinct from,
parathyroid hormone (PTH). PTHRP resembles the native hormone only in its N terminus. It has some
biologic actions similar to those of PTH, and both hormones share a G protein–coupled receptor, known as
PTH/PTHRP receptor (often referred to as PTH-R or PTHRP-R). In contrast to PTH, PTHRP is produced in
small amounts by many normal tissues, including keratinocytes, muscles, bone, and ovary. It regulates
calcium transport in the lactating breast and across the placenta, and seems to regulate development and
remodeling in the lung. Tumors most often associated with paraneoplastic hypercalcemia are carcinomas of
the breast, lung, kidney, and ovary. In breast cancers, PTHRP production is associated with osteolytic bone
disease, bone metastasis, and humoral hypercalcemia. The most common lung neoplasm associated with
hypercalcemia is squamous cell bronchogenic carcinoma. In addition to PTHRP, several other factors, such
as IL-1, TGF-α, TNF, and dihydroxyvitamin D, have also been implicated in causing the hypercalcemia of
malignancy.
The neuromyopathic paraneoplastic syndromes take diverse forms, such as peripheral neuropathies, cortical
cerebellar degeneration, a polymyopathy resembling polymyositis, and a myasthenic syndrome similar to
myasthenia gravis. The cause of these syndromes is poorly understood. In some cases, antibodies,
presumably induced against tumor cell antigens ( Chapter 28 ) that cross-react with neuronal cell antigens,
have been detected. It is postulated that some neural antigens are ectopically expressed by visceral cancers.
For some unknown reason, the immune system recognizes these antigens as foreign and mounts an immune
response.
Acanthosis nigricans is characterized by gray-black patches of verrucous hyperkeratosis on the skin. This
disorder occurs rarely as a genetically determined disease in juveniles or adults ( Chapter 25 ). In addition, in
about 50% of the cases, particularly in those over age 40, the appearance of such lesions is associated with
some form of cancer. Sometimes the skin changes appear before discovery of the cancer.
Hypertrophic osteoarthropathy is encountered in 1% to 10% of patients with bronchogenic carcinomas.
Rarely, other forms of cancer are involved. This disorder is characterized by (1) periosteal new bone
formation, primarily at the distal ends of long bones, metatarsals, metacarpals, and proximal phalanges; (2)
arthritis of the adjacent joints; and (3) clubbing of the digits. Although the osteoarthropathy is seldom seen
in noncancer patients, clubbing of the fingertips may be encountered in liver diseases, diffuse lung disease,
congenital cyanotic heart disease, ulcerative colitis, and other disorders. The cause of hypertrophic
osteoarthropathy is unknown.
Several vascular and hematologic manifestations may appear in association with a variety of forms of
cancer. As mentioned in the discussion of thrombosis , migratory thrombophlebitis (Trousseau syndrome)
may be encountered in association with deep-seated cancers, most often carcinomas of the pancreas or lung.
Disseminated intravascular coagulation may complicate a diversity of clinical disorders ( Chapter 14 ).
Acute disseminated intravascular coagulation is most commonly associated with acute promyelocytic
leukemia and prostatic adenocarcinoma. Bland, small, nonbacterial fibrinous vegetations sometimes form on
the cardiac valve leaflets (more often on left-sided valves), particularly in individuals with advanced mucin-
secreting adenocarcinomas. These lesions, called nonbacterial thrombotic endocarditis, are described
further in Chapter 12 . The vegetations are potential sources of emboli that can further complicate the course
of cancer.

GRADING AND STAGING OF TUMORS
Methods to quantify the probable clinical aggressiveness of a given neoplasm and its apparent extent and
spread in the individual patient are necessary for making an accurate prognosis and for comparing end
results of various treatment protocols. For instance, the results of treating well-differentiated thyroid
adenocarcinoma that is localized to the thyroid gland will be different from those obtained from treating
highly anaplastic thyroid cancers that have invaded the neck organs. Systems have been developed to
express, at least in semiquantitative terms, the level of differentiation, or grade, and extent of spread of a
cancer within the patient, or stage, as parameters of the clinical gravity of the disease.
Grading of a cancer is based on the degree of differentiation of the tumor cells and, in some cancers, the
number of mitoses or architectural features. Grading schemes have evolved for each type of malignancy,
and generally range from two categories (low grade and high grade) to four categories. Criteria for the
individual grades vary with each form of neoplasia and so are not detailed here, but all attempt, in essence,
to judge the extent to which the tumor cells resemble or fail to resemble their normal counterparts. Although
histologic grading is useful, the correlation between histologic appearance and biologic behavior is less than
perfect. In recognition of this problem and to avoid spurious quantification, it is common practice to
characterize a particular neoplasm in descriptive terms, for example, well-differentiated, mucin-secreting
adenocarcinoma of the stomach, or poorly differentiated pancreatic adenocarcinoma. In general, with a few
exceptions, such as soft-tissue sarcomas, grading of cancers has proved of less clinical value than has
staging.
The staging of cancers is based on the size of the primary lesion, its extent of spread to regional lymph
nodes, and the presence or absence of blood-borne metastases. The major staging system currently in use is
the American Joint Committee on Cancer Staging. This system uses a classification called the TNM
system—T for primary tumor, N for regional lymph node involvement, and M for metastases. The TNM
staging varies for each specific form of cancer, but there are general principles. With increasing size the
primary lesion is characterized as T1 to T4. T0 is used to indicate an in situ lesion. N0 would mean no nodal
involvement, whereas N1 to N3 would denote involvement of an increasing number and range of nodes. M0
signifies no distant metastases, whereas M1 or sometimes M2 indicates the presence of metastases and some
judgment as to their number.
LABORATORY DIAGNOSIS OF CANCER
Every year the approach to laboratory diagnosis of cancer becomes more complex, more sophisticated, and
more specialized. For virtually every neoplasm mentioned in this text, the experts have characterized several
subcategories; we must walk, however, before we can run. Each of the following sections attempts to
present the state of the art, avoiding details of method.
Histologic and Cytologic Methods.
The laboratory diagnosis of cancer is, in most instances, not difficult. The two ends of the benign-malignant
spectrum pose no problems; however, in the middle lies a gray zone that the novices dread and where
experts tread cautiously. The focus here is on the roles of the clinician (often a surgeon) and the pathologist
in facilitating the correct diagnosis.
Clinical data are invaluable for optimal pathologic diagnosis, but often clinicians underestimate its value.
Radiation changes in the skin or mucosa can be similar to those associated with cancer. Sections taken from
a healing fracture can mimic an osteosarcoma. Moreover, the laboratory evaluation of a lesion can be only
as good as the specimen made available for examination. It must be adequate, representative, and properly
preserved. Several sampling approaches are available: (1) excision or biopsy, (2) needle aspiration, and (3)
cytologic smears. When excision of a small lesion is not possible, selection of an appropriate site for biopsy
of a large mass requires awareness that the periphery may not be representative and the center largely

necrotic. Appropriate preservation of the specimen is obvious, yet it involves such actions as prompt
immersion in a usual fixative (commonly formalin solution, but other fluids can be used), preservation of a
portion in a special fixative (e.g., glutaraldehyde) for electron microscopy, or prompt refrigeration to permit
optimal hormone, receptor, or other types of molecular analysis. Requesting “quick-frozen section”
diagnosis is sometimes desirable, for example, in determining the nature of a mass lesion or in evaluating
the margins of an excised cancer to ascertain that the entire neoplasm has been removed. This method
permits histologic evaluation within minutes. In experienced, competent hands, frozen-section diagnosis is
highly accurate, but there are particular instances in which the better histologic detail provided by the more
time-consuming routine methods is needed—for example, when extremely radical surgery, such as the
amputation of an extremity, may be indicated. Better to wait a day or two despite the drawbacks, than to
perform inadequate or unnecessary surgery.
Fine-needle aspiration of tumors is another approach that is widely used. The procedure involves aspirating
cells and attendant fluid with a small-bore needle, followed by cytologic examination of the stained smear.
This method is used most commonly for the assessment of readily palpable lesions in sites such as the
breast, thyroid, and lymph nodes. Modern imaging techniques permit extension of the method to lesions in
deep-seated structures, such as pelvic lymph nodes and pancreas. Fine-needle aspiration is less invasive and
more rapidly performed than are needle biopsies. It obviates surgery and its attendant risks. Although it
entails some difficulties, such as small sample size and sampling errors, in experienced hands it is extremely
reliable, rapid, and useful.
Cytologic (Pap) smears provide yet another method for the detection of cancer. This approach is widely
used to screen for carcinoma of the cervix, often at an in situ stage, but it is also used with many other forms
of suspected malignancy, such as endometrial carcinoma, bronchogenic carcinoma, bladder and prostatic
tumors, and gastric carcinomas; for the identification of tumor cells in abdominal, pleural, joint, and
cerebrospinal fluids; and, less commonly, with other forms of neoplasia.
As pointed out earlier, cancer cells have lowered cohesiveness and exhibit a range of morphologic changes
encompassed by the term anaplasia. Thus, shed cells can be evaluated for the features of anaplasia
indicative of their origin from a tumor ( Figs. 7-47 and 7-48 ). In contrast to the histologist's task, judgment
here must be rendered based on the features of individual cells or, at most, a clump of cells, without the
supporting evidence of loss of orientation of one cell to another, and (most importantly) evidence of
invasion. This method permits differentiation among normal, dysplastic, and malignant cells and, in
addition, permits the recognition of cellular changes characteristic of carcinoma in situ. The gratifying
control of cervical cancer is the best testament to the value of the cytologic method.
Although histology and exfoliative cytology remain the most commonly used methods in the diagnosis of
cancer, new techniques are being constantly added to the tools of the surgical pathologist. Some, such as
immunohistochemistry, are already well established and widely used; others, including molecular methods,
are rapidly finding their way into the “routine” category. Only some highlights of these diagnostic
modalities are presented.
Immunohistochemistry.
The availability of specific antibodies has greatly facilitated the identification of cell products or surface
markers. Some examples of the utility of immunohistochemistry in the diagnosis or management of
malignant neoplasms follow.
• Categorization of undifferentiated malignant tumors: In many cases malignant tumors of diverse
origin resemble each other because of limited differentiation. These tumors are often quite difficult to
distinguish on the basis of routine hematoxylin and eosin (H&E)–stained tissue sections. For
example, certain anaplastic carcinomas, lymphomas, melanomas, and sarcomas may look quite
similar, but they must be accurately identified because their treatment and prognosis are different.

Antibodies specific to intermediate filaments have proved to be of particular value in such cases,
because solid tumor cells often contain intermediate filaments characteristic of their cell of origin.
For example, the presence of cytokeratins, detected by immunohistochemistry, points to an epithelial
origin (carcinoma), whereas desmin is specific for neoplasms of muscle cell origin.
• Determination of site of origin of metastatic tumors: Many cancer patients present with metastases.
In some the primary site is obvious or readily detected on the basis of clinical or radiologic features.
In cases in which the origin of the tumor is obscure, immunohistochemical detection of tissue-
specific or organ-specific antigens in a biopsy specimen of the metastatic deposit can lead to the
identification of the tumor source. For example, prostate-specific antigen (PSA) and thyroglobulin
are markers of carcinomas of the prostate and thyroid, respectively.
• Detection of molecules that have prognostic or therapeutic significance: Immunohistochemical
detection of hormone (estrogen/progesterone) receptors in breast cancer cells is of prognostic and
therapeutic value because these cancers are susceptible to anti-estrogen therapy ( Chapter 23 ). In
general, receptor-positive breast cancers have a better prognosis. Protein products of oncogenes such
as ERBB2 in breast cancers can also be detected by immunostaining. Breast cancers with
overexpression of ERBB2 protein generally have a poor prognosis. In general practice, the
overexpression of ERBB2 is confirmed by fluorescent in situ hybridization (FISH) to confirm
amplification of the genomic region containing the ERBB2 gene.
Flow Cytometry.
Flow cytometry can rapidly and quantitatively measure several individual cell characteristics, such as
membrane antigens and the DNA content of tumor cells. Flow cytometry has also proved useful in the
identification and classification of tumors arising from T and B lymphocytes and from mononuclear-
phagocytic cells.
Molecular Diagnosis.
Several molecular techniques—some established, others emerging—have been used for diagnosis and, in
some cases, for predicting behavior of tumors.
• Diagnosis of malignant neoplasms: Although molecular methods are not the primary modality of
cancer diagnosis, they are of considerable value in selected cases. Molecular techniques are useful in
differentiating benign (polyclonal) proliferations of T or B cells from malignant (monoclonal)
proliferations. Because each T and B cell has unique rearrangements of its antigen receptor genes (
Chapter 6 ), PCR–based detection of T-cell receptor or immunoglobulin genes allows distinction
between monoclonal (neoplastic) and polyclonal (reactive) proliferations. Many hematopoietic
neoplasms (leukemias and lymphomas) are associated with specific translocations that activate
oncogenes. Detection of such translocations, usually by routine cytogenetic analysis or by FISH
technique , is often extremely helpful in diagnosis.
[189]
In some cases, molecular techniques, such as
PCR, can detect residual disease in cases that appear negative by conventional analysis. Diagnosis of
sarcomas ( Chapter 26 ) with characteristic translocations is also aided by molecular techniques,
because chromosome preparations are often difficult to obtain from solid tumors. For example, many
sarcomas of childhood, so-called round blue cell tumors ( Chapter 10 ), can be difficult to distinguish
from each other on the basis of morphology. However, the presence of the characteristic
[t(11;22)(q24;q12)] translocation, established by PCR, in one of these tumors confirms the diagnosis
of Ewing sarcoma.A molecular cytogenetic technique called spectral karyotyping has great
sensitivity and allows the examination of all chromosomes in a single experiment.This technique,
which is based on 24-color chromosomal painting with a mixture of fluorochromes, can detect all
types of chromosomal rearrangements in tumor cells, even small, cryptic translocations and
insertions. It can also detect the origin of unidentified chromosomes, called marker chromosomes,
seen in many hematopoietic malignancies. Another available technique is comparative genomic

hybridization, now more conveniently converted to microarray format, which allows the analysis of
chromosomal gains and losses in tumor cells. The use of DNA microarrays (discussed later), either
tiling arrays, which cover the entire human genome, or single-nucleotide polymorphism arrays (SNP
chips), also allows analysis of genomic amplifications and deletions at very high resolution.
• Prognosis of malignant neoplasms: Certain genetic alterations are associated with poor prognosis,
and hence their detection allows stratification of patients for therapy. For example, amplification of
the N-MYC gene and deletions of 1p bode poorly for patients with neuroblastoma, while
amplification of HER-2/NEU in breast cancer is an indication that therapy with antibodies against the
ERBB2 receptor may be effective. These can be detected by routine cytogenetics and also by FISH
or PCR assays. Oligodendrogliomas in which the only genomic abnormality is the loss of
chromosomes 1p and 19q respond well to therapy and are associated with long-term survival when
compared to tumors with intact 1p and 19q but with EGF receptor amplification.
[192]
• Detection of minimal residual disease: After treatment of patients with leukemia or lymphoma, the
presence of minimal disease or the onset of relapse can be monitored by PCR-based amplification of
nucleic acid sequences unique to the malignant clone. For example, detection of BCR-ABL transcripts
by PCR gives a measure of the residual leukemia cells in treated patients with CML. Similarly,
detection of specific KRAS mutations in stool samples of persons previously treated for colon cancer
can alert the clinician to the possible recurrence of the tumor. The prognostic importance of minimal
disease has been established in acute lymphoblastic leukemia, and is being evaluated in other
neoplasms.
• Diagnosis of hereditary predisposition to cancer: As was discussed earlier, germ-line mutations in
several tumor suppressor genes, including BRCA1, BRCA2, and the RET proto-oncogene, are
associated with a high risk of developing specific cancers. Thus, detection of these mutated alleles
may allow the patient and physician to devise an aggressive screening program, consider the option
of prophylactic surgery, and counseling of relatives at risk. Such analysis usually requires detection
of a specific mutation (e.g., RET gene) or sequencing of the entire gene. The latter is necessitated
when several different cancer-associated mutations are known to exist. Although the detection of
mutations in such cases is relatively straightforward, the ethical issues surrounding such
presymptomatic diagnosis are complex.
Tumor Markers
Biochemical assays for tumor-associated enzymes, hormones, and other tumor markers in the blood cannot
be used for definitive diagnosis of cancer; however, they contribute to the detection of cancer and in some
instances are useful in determining the effectiveness of therapy or the appearance of a recurrence.
A host of tumor markers have been described, and new ones are identified every year. Only a few have
stood the test of time and proved to have clinical usefulness.
The application of several markers, listed in Table 7-12 , is considered in the discussion of specific forms of
neoplasia in other chapters, so only a few widely used examples suffice here. PSA, used to screen for
prostatic adenocarcinoma, may be one of the most used, and most successful, tumor markers in clinical
practice. Prostatic carcinoma can be suspected when elevated levels of PSA are found in the blood.
However, PSA screening also highlights problems encountered with virtually every tumor marker. Although
PSA levels are often elevated in cancer, PSA levels also may be elevated in benign prostatic hyperplasia .
Furthermore, there is no PSA level that ensures that a person does not have prostate cancer. Thus, the PSA
test suffers from both low sensitivity and low specificity. Other tumor markers occasionally used in clinical
practice include CEA, which is elaborated by carcinomas of the colon, pancreas, stomach, and breast, and
AFP, which is produced by hepatocellular carcinomas, yolk sac remnants in the gonads, and occasionally
teratocarcinomas and embryonal cell carcinomas. Unfortunately, like PSA, both of these markers can be
produced by a variety of non-neoplastic conditions as well. Thus, as with PSA levels, CEA and AFP assays
lack both specificity and sensitivity required for the early detection of cancers. They are still useful in the
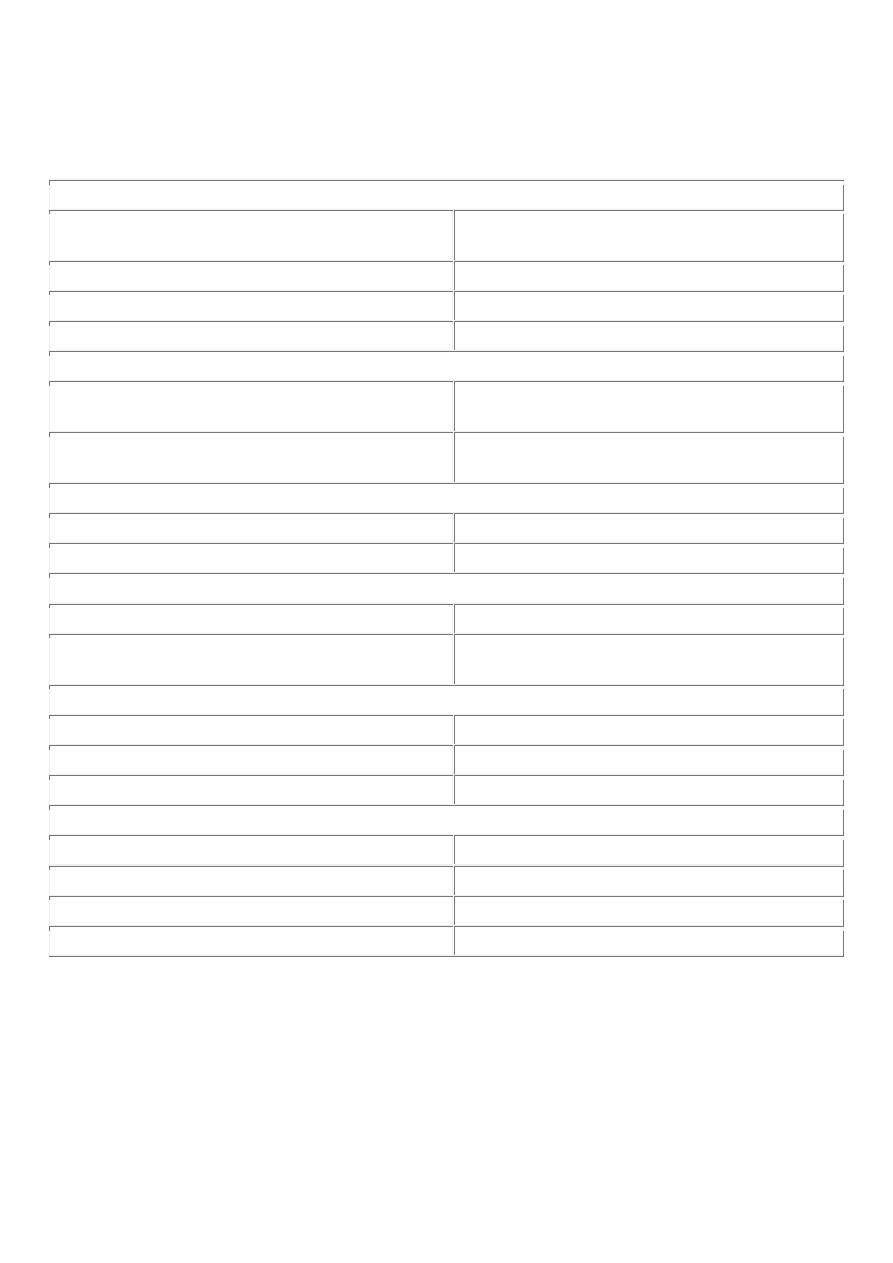
detection of recurrences after excision. With successful resection of the tumor, these markers disappear from
the serum; their reappearance almost always signifies the beginning of the end.
TABLE 7-12 -- Selected Tumor Markers
HORMONES
Human chorionic gonadotropin
Trophoblastic tumors, nonseminomatous testicular
tumors
Calcitonin
Medullary carcinoma of thyroid
Catecholamine and metabolites
Pheochromocytoma and related tumors
Ectopic hormones
See “Paraneoplastic Syndromes” ( Table 7-11 )
ONCOFETAL ANTIGENS
α-Fetoprotein
Liver cell cancer, nonseminomatous germ cell
tumors of testis
Carcinoembryonic antigen
Carcinomas of the colon, pancreas, lung, stomach,
and heart
ISOENZYMES
Prostatic acid phosphatase
Prostate cancer
Neuron-specific enolase
Small-cell cancer of lung, neuroblastoma
SPECIFIC PROTEINS
Immunoglobulins
Multiple myeloma and other gammopathies
Prostate-specific antigen and prostate-specific
membrane antigen
Prostate cancer
MUCINS AND OTHER GLYCOPROTEINS
CA-125
Ovarian cancer
CA-19-9
Colon cancer, pancreatic cancer
CA-15-3
Breast cancer
NEW MOLECULAR MARKERS
p53, APC, RAS mutants in stool and serum
Colon cancer
p53 and RAS mutants in stool and serum
Pancreatic cancer
p53 and RAS mutants in sputum and serum
Lung cancer
p53 mutants in urine
Bladder cancer
Other widely used markers include human chorionic gonadotropin for testicular tumors, CA-125 for ovarian
tumors, and immunoglobulins in multiple myeloma and other secretory plasma cell tumors. The
development of tests to detect cancer markers in blood and body fluids is an active area of research. Some of
the markers being evaluated include the detection of mutated APC, p53, and RAS in the stool of individuals
with colorectal carcinomas; the presence of mutated p53 and of hypermethylated genes in the sputum of
persons with lung cancer and in the saliva of persons with head and neck cancers; and the detection of
mutated p53 in the urine of patients with bladder cancer.