Pediatric urology
Dr.Mohammed BassilUndescended testes
The testes descend into the scrotum in the third trimester (passing through the inguinal canal at 24–28 weeks). Failure of testicular descent results in cryptorchidism (or undescended testes).Incidence
Incidence is 3% at birth (unilateral > bilateral). Approximately 80% will spontaneously descend by 3 months. The incidence at 1 year is 1%.
Classification
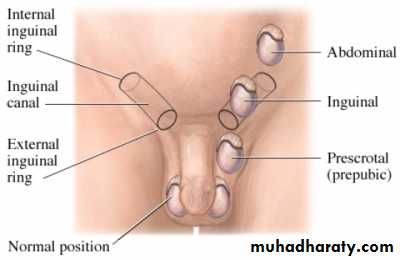
• Retractile: an intermittent active cremasteric reflex causes the testis to retract up and out of the scrotum.• Ectopic (<5%): abnormal testis migration below the external ring of the inguinal canal (to perineum, base of penis, or femoral areas)
• Incomplete descent (~95%): testis may be intra-abdominal, inguinal,or prescrotal
• Atrophic/absent
Risk factors
These include preterm infants, low birth weight, small for gestational age,
and twins.
Etiology
This includes abnormal testis or gubernaculum (tissue that guides the testis into the scrotum during development); endocrine abnormalities (low level of androgens, human chorionic gonadotrophin [hCG], luteinizing hormone (LH), calcitonin gene–related peptide); and decreased intraabdominal pressure (prune-belly syndrome, gastroschisis).
Pathology
There is degeneration of Sertoli cells, loss of Leydig cells, and atrophy and
abnormal spermatogenesis.
Long-term complications
• Relative risk of cancer is 40-fold higher in the undescended testis. Most are seminomas; carcinoma in situ represents a small percentage (~2%).There is a slightly increased risk of cancer in the contralateral, normally descended testis.• Reduced fertility
• Increased risk of testicular torsion
• Increased risk of direct inguinal hernias (due to a patent processus vaginalis)
Management
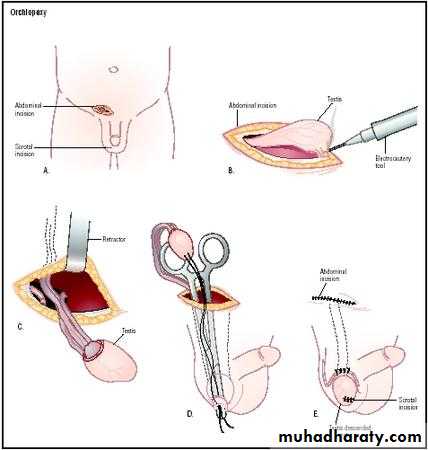
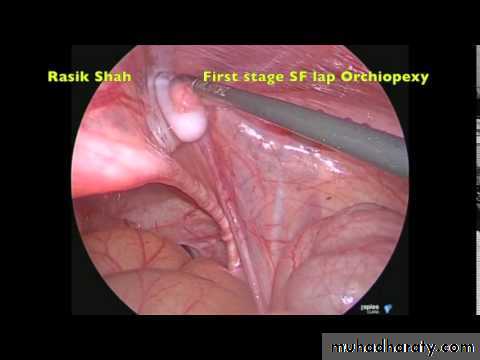
Full examination is required to elucidate if the testis is palpable and to identify location. Assess for associated congenital defects. If neither testis is palpable, consider chromosome analysis (to exclude an androgenized female) and hormone testing (high LH and FSH with a low testosterone indicates anorchia).Treatment should be performed within the first year. Hormone therapy (hCG, LHRH) stimulates testosterone production. Surgery consists of inguinal exploration, mobilization of spermatic cord, ligation of processus vaginalis, and securing the testis into a dartos pouch in the scrotal wall (orchidopexy). Laparoscopy can be used in planning surgery and for treatment. Intra-abdominal testes may require division of spermatic vessels to provide extra length (Fowler-Stevens procedure, relying on collateral
blood flow from vas), two-stage procedures, or microvascular
autotransplantation.
Pelviureteric junction (PUJ)obstruction
A ureteropelvic junction (UPJ) obstruction can be thought of as a restriction to flow of urine, from the renal pelvis to the ureter, which, if left uncorrected, will lead to progressive renal deterioration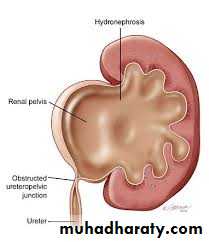
UPJ obstruction occurs in all pediatric age groups, but there tends to be a clustering in the neonatal period because of the detection of antenatal hydronephrosis and again later in life because of symptomatic occurrence. At one point about 25% of cases were discovered within the first year of life.
UPJ obstruction is the most common cause of significant dilation of the collecting system in the fetal kidney.
Pelviureteric junction (UPJ)obstruction
Pelviureteric junction (PUJ)obstruction
Obstruction occurs more commonly in boys than in girls.Left-sided lesions predominate, particularly in the neonate (approximately 67%).
Bilateral UPJ obstruction is present in 10% to 40% of cases.
and it has been known to affect members of more than one generation.
ETIOLOGY
Intrinsic:
interruption in the development of the circular musculature of the UPJ.
or an alteration of the collagen fibers and composition between and around the muscle cells (The muscle fibers become widely separated and attenuated, leading to a functional discontinuity of the muscular contractions and ultimately to insufficient emptying) .
a significant increase in the lamina muscularis and in the number of inner longitudinal muscular bundles of the UPJ complex of obstructed kidneys in infants younger than 1 year of age, compared with age-matched normal infants
ETIOLOGY
valvular mucosal folds.persistent fetal convolutions .
upper ureteral polyps.
Such folds that does not flatten out when the ureter is distended or stretched.
"östling's folds" are now considered folds that are not obstructive and disappear with a person's lineargrowth they are rarely seen in an older child or adult.ETIOLOGY
Extrinsic:An aberrant, accessory, or early-branching lower-pole vessel is the most common cause of extrinsic UPJ obstruction. These vessels pass anteriorly to the UPJ or proximal ureter and contribute to mechanical obstruction.
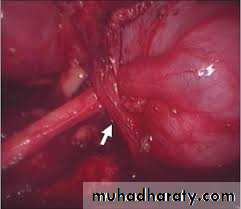
ETIOLOGY
an aberrant or accessory renal artery to the lower pole of the kidney is present and the ureter courses behind it, the ureter may angulate at both the UPJ and the point at which it traverses over the vessel as the pelvis fills and bulges anteriorly. Further angulation of the ureter occurs as it becomes adherent to the UPJ secondary to an inflammatory process. A two-point obstruction ensues, with kinking of the ureter at the UPJ and at the point where the ureter drapes over the vessel.
Secondary Ureteral Pelvic Junction Obstruction
UPJ obstruction may also be seen with severe vesicoureteral reflux (VUR); these conditions coexist in 10% of cases. The ureter elongates and develops a tortuous course in response to the obstructive element of reflux. A kink may develop in the UPJ area, a point of relative fixation, and may cause obstruction secondarily . In such a situation the obstructive lesion needs to be corrected initially, even though the VUR contributed to the initial problem.Associated Anomalies
UPJ obstruction is the most common anomaly encountered in the opposite kidney; it occurs in 10% to 40% of cases.Renal dysplasia and multicystic dysplastic kidney are the next most frequently observed contralateral lesions.
In addition, unilateral renal agenesis has been noted in almost 5% of children.
UPJ obstruction may also occur in either the upper or the lower half (usually the latter) of a duplicated collecting system,or of a horseshoe or ectopic kidney.
VUR has been found in as many as 40% of affected children.
UPJ obstruction was noted in 21% of children with the VATER (vertebral defects, imperforate anus, tracheoesophageal fistula, and radial and renal dysplasia) association.
PRESENTATION
most infants are asymptomatic and most children are discovered because of their symptoms. infants were discovered to have UPJ obstruction because of a palpable mass.infants who present with failure to thrive, feeding difficulties, sepsis secondary to urinary tract infection, or pain or hematuria related to nephrolithiasis. Urinary tract infection is the presenting sign in 30% of affected children beyond the neonatal period.
PRESENTATION
In the older child, episodic flank or upper abdominal pain, sometimes associated with nausea and vomiting due to intermittent UPJ obstruction, is a prominent symptom.cyclic vomiting alone is caused by intermittent UPJ obstruction.
Hematuria, which is seen in 25% of children, may occur after minor abdominal trauma.
This hematuria is believed to be caused by disruption and rupture of mucosal vessels in the dilated collecting system.
PRESENTATION
In the young adult, episodic flank or abdominal pain, particularly during diuresis, is a common manifestation. Occasionally, a patient with the UPJ obstruction presents with hypertension. The pathophysiology is thought to be a functional ischemia with reduced blood flow caused by the enlarged collecting system that produces a renin-mediated hypertension .
DIAGNOSIS
Ultrasonography; is the standard method for identifying hydronephrosis in infancy. Postnatal ultrasound imaging is usually deferred until day 3 of life, to allow for improvement in the relative oliguria, which could lead to underestimation of the degree of hydronephrosis.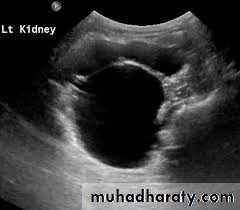
DIAGNOSIS
If prenatal US has shown a large or bilateral hydronephrosis, a follow-uprenal tract ultrasound scan should be performed soon after birth. If there
is a prenatal unilateral hydronephrosis (and the bladder is normal), the
scan is deferred until days 3–7 (to allow normal physiological diuresis to
occur, which may spontaneously improve or resolve hydronephrosis).
If upper tract obstruction persists, a voiding cystourethrogram (VCUG)
is indicated (to rule out VUR and examine for posterior urethral valves),
and a renogram can assess individual renal function and drainage (DTPA,
MAG-3).
DIAGNOSIS
IVU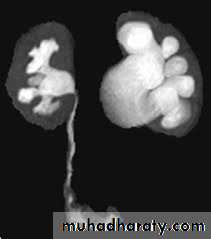
DIAGNOSIS
CT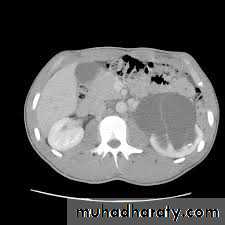
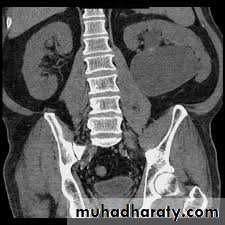
Treatment
Children may be observed with US and renogram if they remain stable andhave good renal function and no other complications (such as persistent infection or stones).
If children are symptomatic or have a signifi cant hydronephrosis with impaired renal function (<40%), pyeloplasty is recommended
PYELOPLASTY
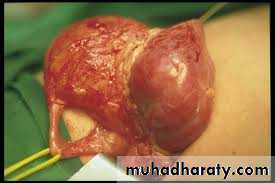
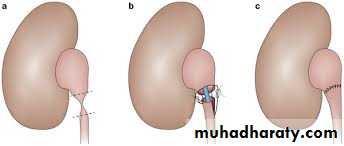
Vesicoureteric reflux (VUR)
VUR results from abnormal retrograde flow of urine from the bladder into the upper urinary tract.Epidemiology
Overall incidence in children is >10%, with younger children affected more than older children, girls more than boys (female–male ratio 5:1). VUR occurs more often in Caucasian than in Afro-Caribbean children.
Siblings of an affected child have a 40% risk of reflux, and routine screening of siblings is recommended.
Pathogenesis
The ureter passes obliquely through the bladder wall (1–2 cm), where it is supported by muscular attachments that prevent urine reflux during bladder filling and voiding. The normal ratio of intramural ureteric length to ureteric diameter is 5:1. Reflux occurs when the intramural length of ureter is too short (ratio<5:1). The degree of reflux is graded I–V . The appearance of the ureteric orifice changes with increasing severity of reflux, classically described as stadium, horseshoe, golf-hole, or patulous.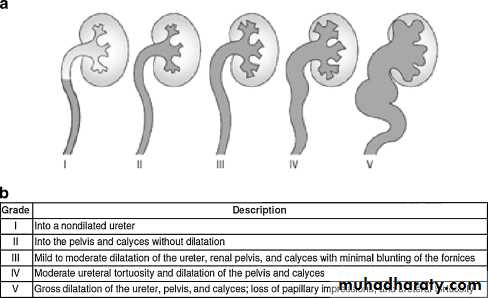
Classification
• Primary reflux (1%) results from a congenital abnormality of theureterovesical junction.
• Secondary refl ux results from urinary tract dysfunction associated
with elevated intravesical pressures. Causes include posterior urethral
valves (reflux seen in 50%), urethral stenosis, neuropathic bladder, and
detrusor sphincter dyssynergia (DSD).
• VUR is also seen with duplex ureters. The Weigert–Meyer rule states
that the lower-pole ureter enters the bladder proximally and laterally,
resulting in a shorter intramural tunnel, which predisposes to reflux.
Complications
VUR associated with UTI can result in refl ux nephropathy with hypertension and progressive renal failure.
Presentation
Patients have symptoms of UTI, fever, dysuria, suprapubic or abdominal pain, failure to thrive, vomiting, and diarrhea.
Investigation
• Urinalysis and culture to diagnose UTI
• Urinary tract ultrasound scan and VCUG to diagnose and grade reflux and establish reversible causes .
• Urodynamic assessment if suspicious of voiding dysfunction
• DMSA scan to detect and monitor associated renal cortical scarring.
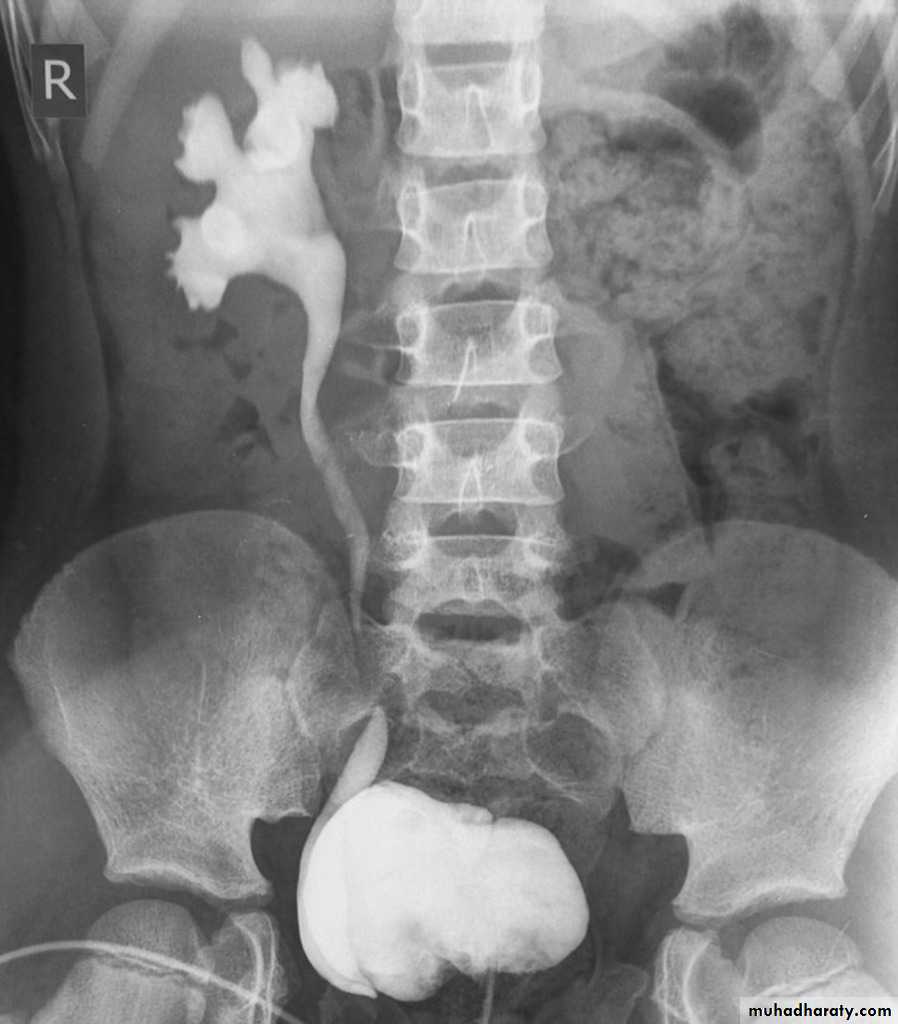
Management
Correct problems contributing to secondary reflux. Most primary VUR grade I–II cases will resolve spontaneously (~85%), with 50% resolution in grade III. Observation and medical treatment are initially recommended.Medical treatment
Low-dose antibiotic prophylaxis should be given to keep the urine sterile and lower the risk of renal damage until refl ux resolves. Anticholinergic drugs are given to treat bladder overactivity.
Typical indications for antireflux surgery include
• Breakthrough UTIs despite prophylactic antibiotics• Noncompliance with medical management
• Severe grades of reflux (grade IV or V), especially with pyelonephritic changes
• Failure of renal growth, new renal scars, or deterioration of renal function on serial ultrasounds or scans
• Reflux that persists in girls as full linear growth is approached at puberty
• Reflux associated with congenital abnormalities at the UVJ (e.g., bladder diverticula)
Surgery is indicated for severe refl ux, breakthrough UTIs, evidence of
progressive renal scarring, and VUR that persists after puberty.
Techniques
of ureteral re-implantation include the following:
• Intravesical methods involve mobilizing the ureter and advancing it
across the trigone (Cohen repair) or reinsertion into a higher, medial
position in the bladder (Politano–Leadbetter repair).
• Extravesical techniques involve attaching the ureter into the bladder
base and suturing muscle around it (Lich–Gregoir procedure).
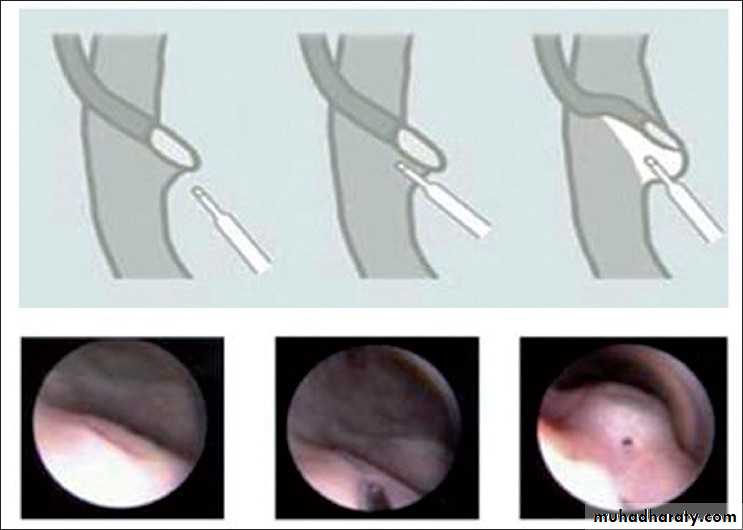
• Alternatively, endoscopic subtrigonal injection of Defl ux into the
ureteral orifi ce has 70% success, and 95% with repeated treatments.
MEGAURETEr
Definition
MGU is a nonspecific term implying a spectrum of anomalies associated with pathologically excessive ureteral diameter
Children up to 12 year-ureteral width>7mm
Older children ureteral width>10mm
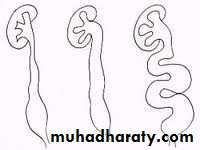
Classification
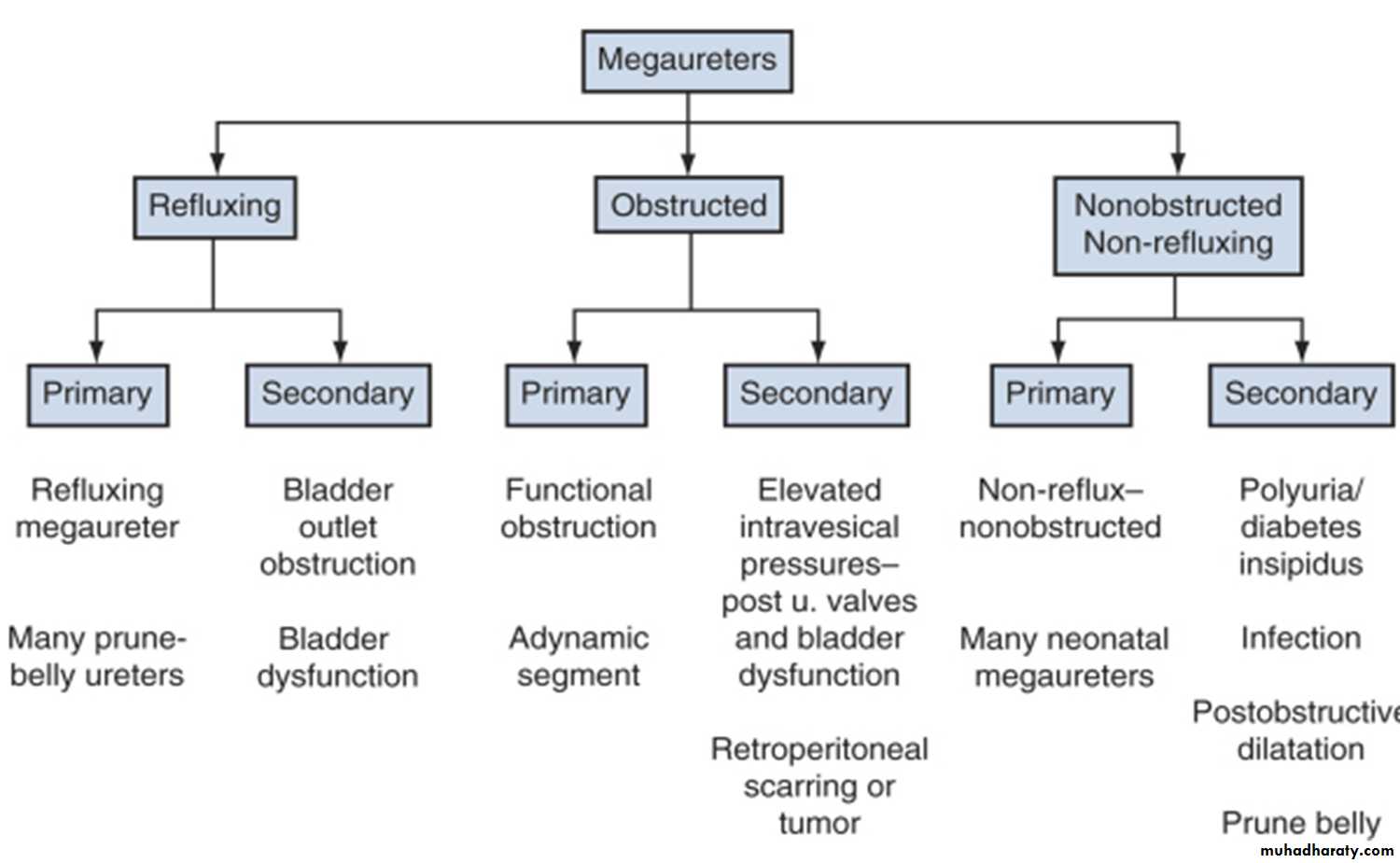
presentation
Bilateral in approximately 25% of patients .In up to 10% to 15% of children the contralateral kidney may be absent or dysplastic .
UTIs, abdominal pain, or hematuria.
The diagnosis may be made later in life in some asymptomatic patients.
Pathophysiology
The distal end of the ureter, as it becomes intramural and subsequently submucosal, rearranges the muscular layers in its wall. All layers become longitudinally oriented, and the ureteral adventitia fuses to the bladder trigone .Primary Obstructive Megaureter
the precise etiology of primary obstructive MGU remains unclear, it is generally agreed that the most common finding is an aperistaltic juxtavesical (adynamic) ureteral segment that prevents urine from flowing at an acceptable rate. , and functional obstruction results
Operative exposure a dynamic segment
TREATMENT
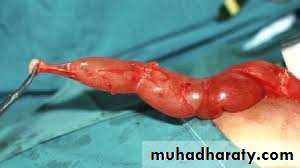
Penile and urethral disorders
Dr.Mohammed Bassilcongenital deformity in which the opening of the urethra occurs on the underside part of the penis, anywhere from the glans to the perineum. It is often associated with a hooded foreskin and chordee.
It occurs in 1 in 250 live male births. There is an 8% incidence in offspring of an affected male, and a 14% risk in male siblings.
Hypospadias
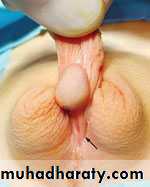
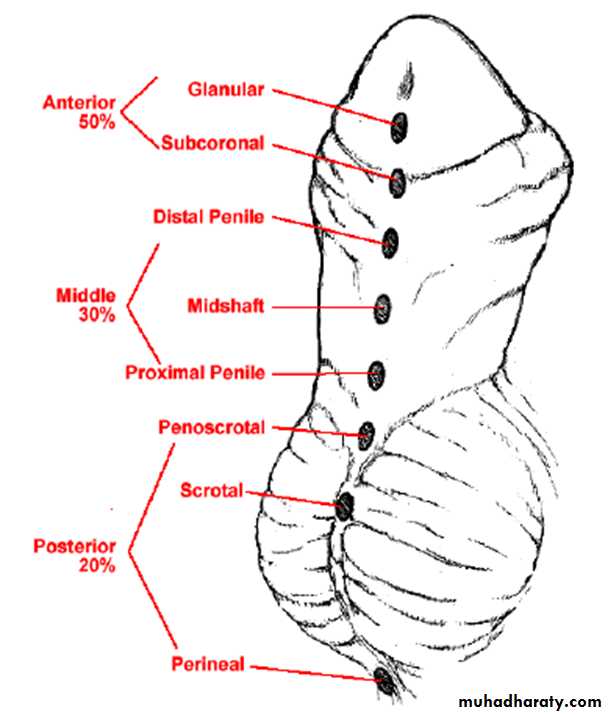
Classifi cation
Hypospadias can be classified according to the anatomical location of the urethral meatus .Anterior (or distal)—glandular, coronal, and subcoronal (~50%)
Middle—distal penile, midshaft, and proximal penile (~30%)
Posterior (or proximal)—penoscrotal, scrotal, and perineal (~20%)
Hypospadias
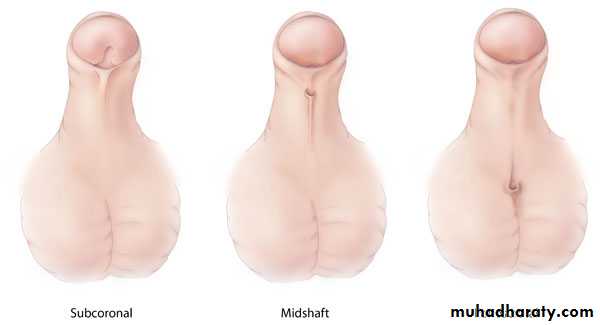
Etiology
Hypospadias results from incomplete closure of urethral folds on the underside of the penis .Chordee is caused by abnormal urethral plate development.
and the hooded foreskin is due to failed formation of the glandular urethra and fusion of the preputial folds.
Hypospadias
A full clinical examination will make the diagnosis. However, it is also important to seek out associated abnormalities that will need treatment.
Patients with absent testes and severe hypospadias should undergo chromosomal and endocrine investigation to exclude intersex conditions.
Hypospadias
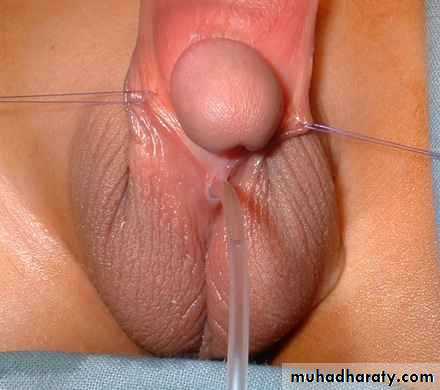
Treatment
Surgery is indicated where deformity is severe, interferes with voiding, OR is predicted to interfere with sexual function. Surgery is now performed between 6 and 12 months of age. The aim of surgery is to correct penile curvature (orthoplasty), reconstruct a new urethra, and bring the new meatus to the tip of the glans.Severe cases may require staged procedures.Hypospadias
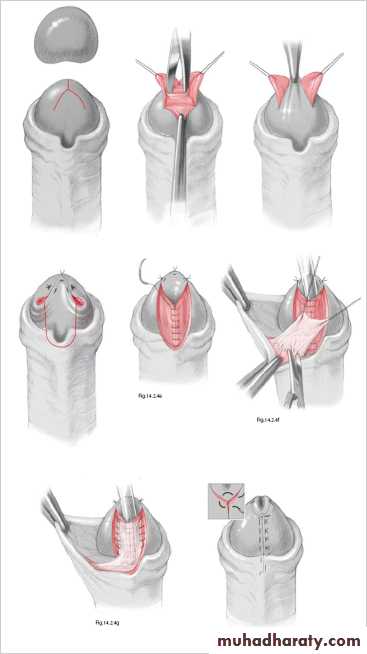

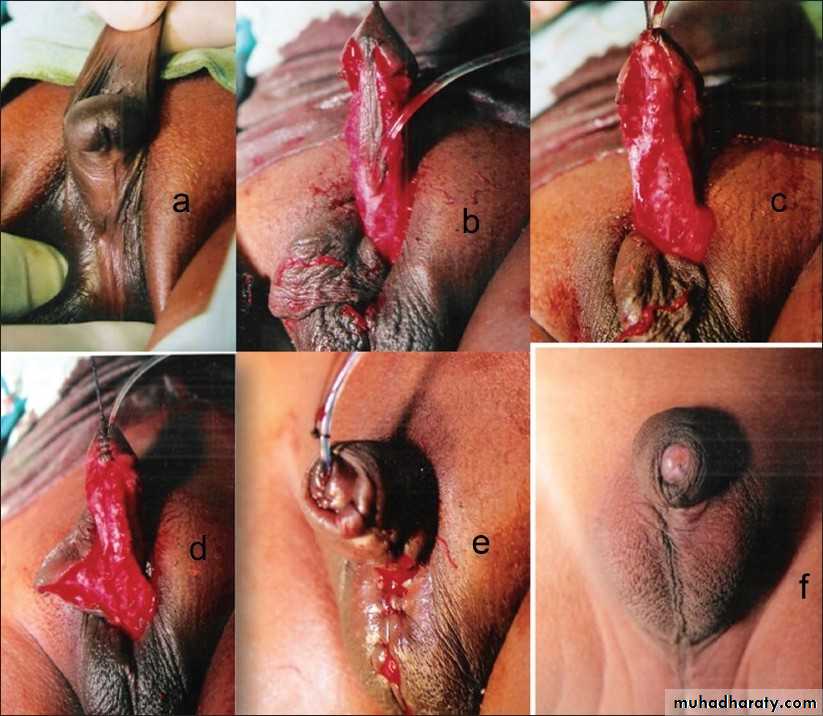
In epispadias, the urethra opens onto the dorsal surface of the penis, anywhere from the glans, penile shaft, or, most commonly, the penopubic region. An incomplete urethral sphincter mechanism results in a high risk of incontinence.
Epispadias is also associated with dorsal chordee , with incomplete foreskin dorsally. Epispadias is part of the exstrophy–epispadias complex
Epispadias
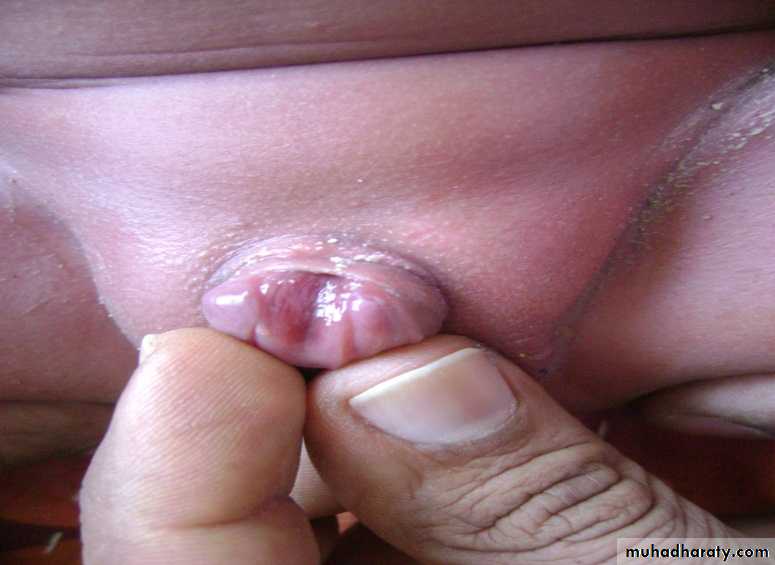
Associated anomalies
Diastasis of the symphysis pubis results in splaying of the corpora cavernosa and shortening of the penile shaft. Females have a bifid clitoris and poorly developed labia and demonstrate a spectrum of urethral deformities ranging from a patulous urethral orifice to a urethral cleft affecting the entire length of the urethra and sphincter.There is a 40% risk of vesicoureteric reflux (VUR).
Epispadias
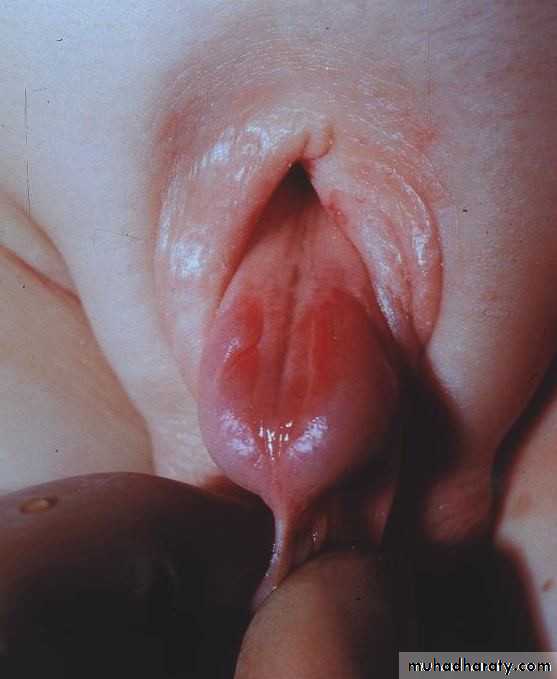
Incidence
Epispadias affects 1 in 117,000 males. It is rarely seen in females (male–female ratio is 5:1).Treatment
This involves urethroplasty with functional and cosmetic reconstruction of the external genitalia (penile lengthening and correction of chordee)at 6–12 months.
From age 4–5 years, when children can be toilet trained, bladder neck reconstruction can be performed . This achieves continence, and any bladder residuals may then be emptied by urethral catheterization. If this surgery fails, insertion of artifi cial urinary sphincters .
Epispadias
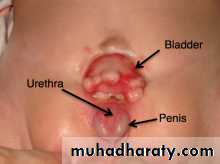
Posterior urethral valves (PUV) are abnormal congenital mucosal folds in the prostatic (posterior) urethra causing lower urinary tract obstruction.
Classification
Type I (90–95%): Membranes arise from the distal lateral aspect of the verumontanum,1 which extend distally and anteriorly to fuse in the midline.
Type II: Longitudinal folds extending from the verumontanum to bladder neck .
Type III (5%): A ring-like membrane found distal to the verumontanum
Incidence is 1 in >5000 males.
Posterior urethral valves
EtiologyNormal male urethra has small, paired lateral folds found between the lateral, distal edge of verumontanum and lateral urethral wall.
PUVs probably represent a congenital overgrowth of these folds from abnormal insertion of Wolffi an ducts into the posterior urethra during fetal development.
Posterior urethral valves
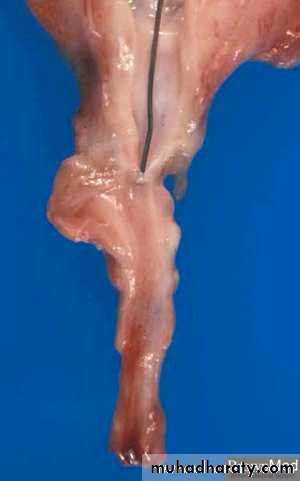
Presentation
Prenatal US features These include bilateral hydroureteronephrosis, dilated bladder with elongated ectatic posterior urethra, thick-walled bladder, oligohydramnios, and renal dysplasia.Newborn and infants
These children have respiratory distress,palpable abdominal mass (hydronephrotic kidney or distended bladder), ascites, UTI, electrolyte abnormalities, and failure to thrive.
Posterior urethral valves
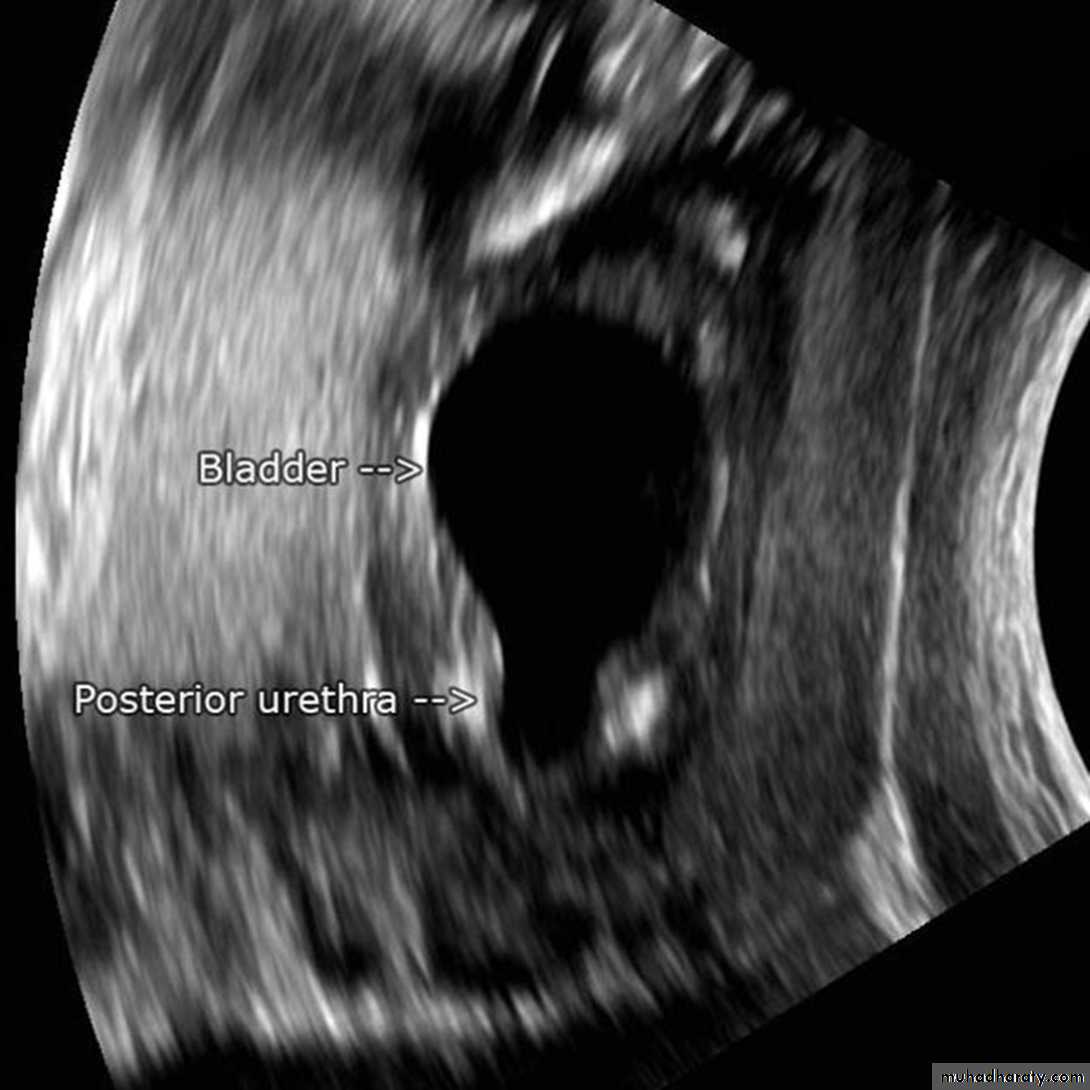

Older children
Milder cases may present later with recurrent UTI, poor urinary stream,incomplete bladder emptying, poor growth, and incontinence. There is arisk of renal failure, vesicoureteric reflux, and voiding dysfunction (overactive or underactive bladder), also described as valve bladder syndrome.Posterior urethral valves
Associated featuresPOP-off valve syndrome is seen in 20%. It describes mechanisms by which high urinary tract pressure is dissipated to allow normal renal development. It includes leaking of urine from small bladder or renal pelvis ruptures (urinary ascites), reflux into a nonfunctioning kidney (vesicoureteral reflux with renal dysplasia [VURD]), and formation of bladder diverticuli.
Posterior urethral valves
InvestigationUltrasound scan of kidneys and bladder.
VCUG shows distended and elongated posterior urethra; partially filled anterior urethra; bladder neck hypertrophy; lucencies representing valve leaflets; thick-walled bladder (±diverticuli); incomplete bladder emptying; reflux (50%).
Isotope renal scan (MAG-3, DMSA) assesses renal function.
Videourodynamics allows diagnosis of associated voiding dysfunction.
Posterior urethral valves
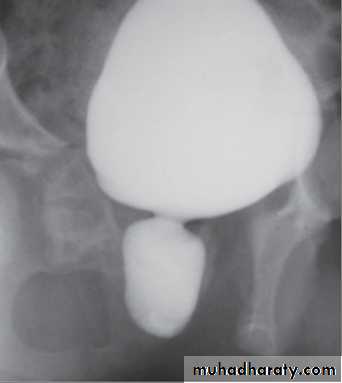
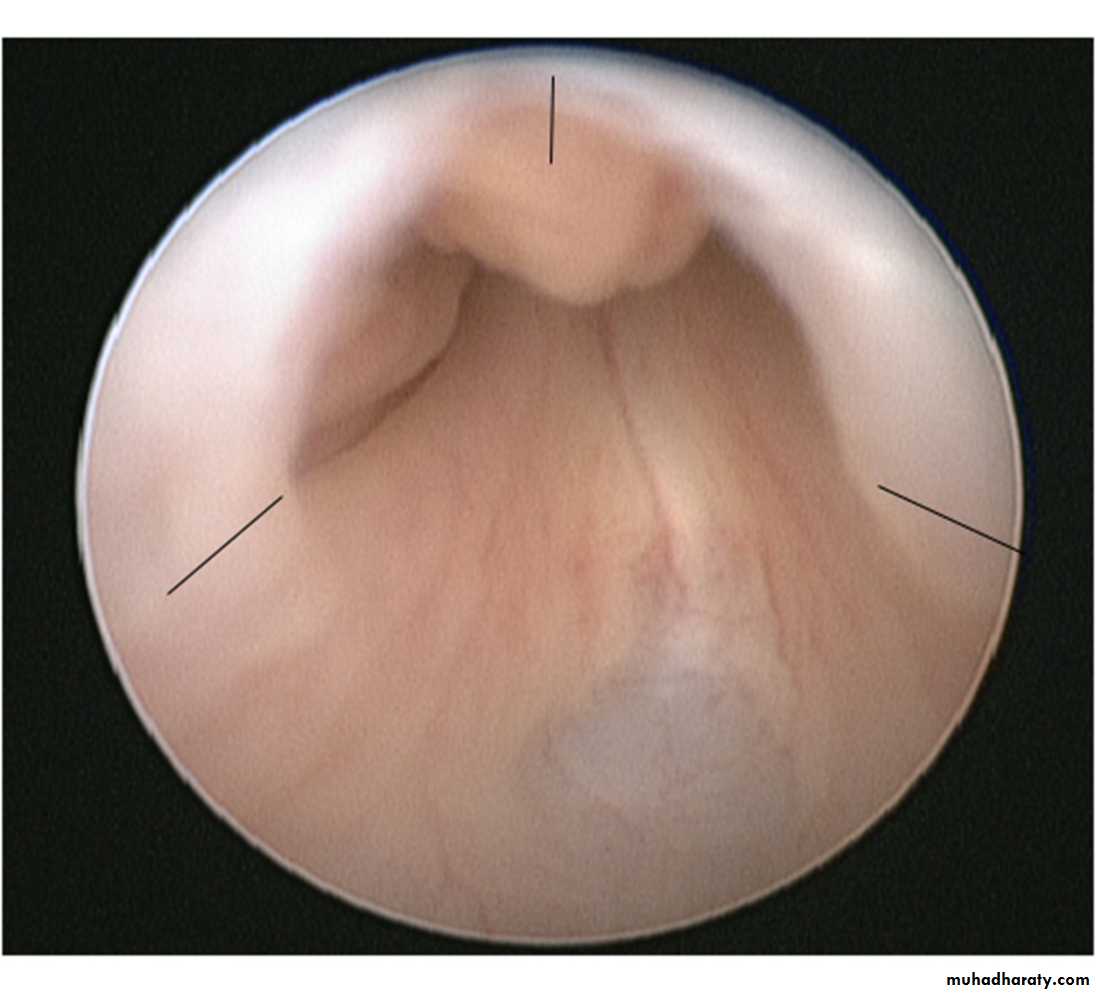
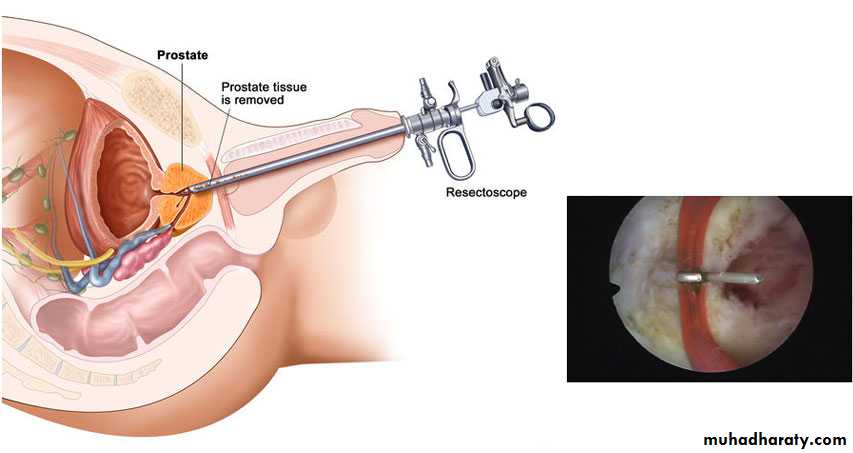
Management
Commence prophylactic antibiotics immediately, check serum electrolytes, and drain the bladder with a pediatric feeding tube. If there is improvement, cystoscopy and transurethral ablation of valve If upper tracts remain dilated with raised creatinine after bladder drainage, a temporary cutaneous vesicostomy is indicated.An alternative is ureterostomy drainage. Valve ablation is performed at a later stage.Prognosis
Prognosis is 35% of patients will have poor renal function; 20% develop end-stage renal failure.