
URINARY SYSTEM
Part 2
OBJECTIVES:
Cystic diseases of the kidney: including
o Autosomal dominant polycystic kidney disease
o Autosomal recessive polycystic kidney disease
o Medullary cystic kidney disease
Listing the causes and pathogenetic factor of an important cause of
urinary outflow obstruction which is the nephrolithiasis and describing
features and complication of its important complication which is the
hydronephrosis.
Listing the pathological features of renal cell carcinoma and its variants.
Listing the pathological features of Wlim's tumor.
Listing and describing the pathological features of ureteric, vescical, and
urethral disorders including:
o Congenital abnormalities
o Inflammatory disorders
o tumors
CYSTIC DISEASES OF THE KIDNEY
These are a heterogeneous group comprising
a. hereditary b. developmental but nonhereditary c. acquired disorders.
They are important for several reasons:
1. They are practically common and often present diagnostic problems
2. Some are major causes of chronic renal failure (adult polycystic disease)
3. They can occasionally be confused clinically with malignant tumors.
4.
Simple Cysts: are a common but have no clinical significance. They can be multiple
or single, commonly up to 5 cm in diameter. They are translucent; filled with clear
fluid; lined by a single layer of cuboidal or flattened epithelium.
Dialysis-associated acquired cysts: occur with prolonged dialysis in those with end-
stage renal disease. They may bleed, causing hematuria. Occasionally, renal
adenomas or carcinomas arise in the walls of these cysts.
Autosomal Dominant (Adult) Polycystic Kidney Disease (ADPKD): is
characterized by multiple expanding cysts of both kidneys that ultimately destroy the
intervening parenchyma. ADPKD is responsible for 10% of all chronic renal failures.
It is caused by inheritance of one of two autosomal dominant genes of very high
penetrance. The kidneys may be very large (up to 4 kg for each), and thus are readily
palpable abdominally. Grossly the kidney is composed of a mass of cysts of varying
sizes (up to 4 cm). The cysts are filled with fluid (clear, turbid, or hemorrhagic).
Microscopically, the cysts have often atrophic, lining. The pressure of the expanding
cysts leads to ischemic atrophy of the intervening renal substance.
Clinically: ADPKD in adults usually does not produce symptoms until the fourth
decade, by which time the kidneys are quite large. Intermittent gross hematuria
commonly occurs. The most important complications are hypertension and urinary
infection.
Saccular aneurysms of the circle of Willis are present in up to 30% of patients, and
these individuals have a high incidence of subarachnoid hemorrhage. Asymptomatic
liver cysts occur in one-third of patients.
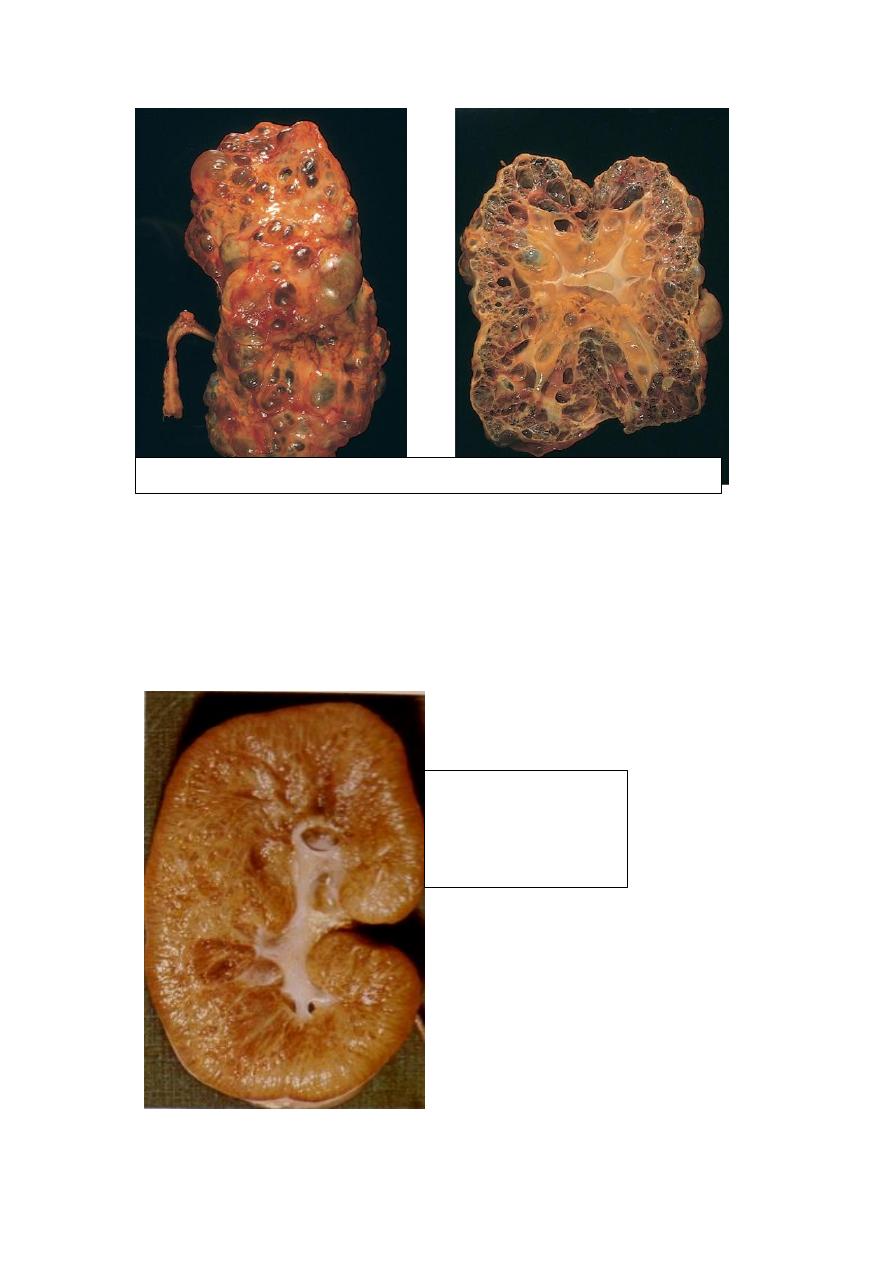
Autosomal Recessive (Childhood) Polycystic Kidney Disease (ARPKD) is a rare
developmental anomaly that is genetically distinct from ADPKD. Perinatal, neonatal,
infantile, and juvenile subcategories have been defined, depending on time of
presentation and the presence of associated hepatic lesions. Both kidneys are
invariably involved with numerous small cysts that give them a sponge-like
appearance. The cysts are lined by cuboidal cells. ARPKD is associated with multiple
epithelium-lined cysts in the liver. Young infants may die quickly from hepatic or
renal failure
.
Autosomal Dominant (Adult) Polycystic Kidney Disease
Autosomal Recessive
(Childhood) Polycystic
Kidney Disease
(ARPKD)

Medullary Cystic Disease (MCD)
This is of two major types of
1. Medullary sponge kidney a relatively common and usually harmless condition and
2. Nephronophthisis-medullary cystic disease complex, which is associated with renal
dysfunction (within 5-10 years). On the basis of the time of onset they are divided
into, infantile, juvenile (the most common), adolescent, and adult types.
Pathologic features of medullary cystic disease include small contracted kidneys with
numerous small typically at the cortico-medullary junction
URINARY OUTFLOW OBSTRUCTION
Renal Stones (Urolithiasis) these are frequent (1% of all autopsies) and mostly form
in the kidney. The majority (80%) are composed of either calcium oxalate or calcium
oxalate mixed with calcium phosphate; 10% are of magnesium ammonium phosphate,
and the remainder 10% are either uric acid or cystine stones. The most important
cause is increased urine concentration of the stone's constituents (supersaturation).
Those who develop calcium stones have
1. Hypercalciuria not associated with hypercalcemia due to either
a. Absorption of calcium from the gut in excessive amounts (i.e. absorptive
hypercalciuria)
b. Primary renal defect of calcium reabsorption (renal hypercalciuria).
Hypercalcemia is present in only 5% to 10% (due to hyperparathyroidism, vitamin D
intoxication, or sarcoidosis) and consequent hypercalciuria. In 20% of this subgroup,
there is excessive excretion of uric acid in the urine, which favors calcium stone
formation; the urates provide a nidus for calcium deposition. A high urine pH favors
crystallization of calcium phosphate and stone formation. Magnesium ammonium
phosphate stones almost always occur in persistently alkaline urine due to UTIs,
particularly with urea-splitting bacteria (as Proteus vulgaris and the Staphylococci).
Medullary
Cystic Disease
(MCD)

Gout and diseases involving rapid cell turnover (as the leukemias), lead to high uric
acid levels in the urine and uric acid stones. About half of the individuals with uric
acid stones, however, have no hyperuricemia but unexplained tendency to excrete
persistently acid urine (under pH 5.5), which favors uric acid stone formation (cf.
stones containing calcium phosphate).
Common sites of formation are renal pelvis and calyces as well as the bladder. Stone
may be small with smooth or jagged surface. Occasionally, progressive accumulation
of salts leads to the development of branching structures known as staghorn calculi,
which create a cast of the renal pelvis and calyceal system. These massive stones are
usually composed of magnesium ammonium phosphate; these do not produce
symptoms or significant renal damage. Smaller stones may pass into the ureter,
producing a typical intense pain known as renal colic. Often at this time there is gross
hematuria. The clinical significance of stones lies in their capacity to obstruct urine
flow or to produce sufficient trauma to cause ulceration and bleeding. In either case,
they predispose the sufferer to bacterial infection.
Hydronephrosis refers to dilation of the renal pelvis and calyces, with accompanying
atrophy of the renal parenchyma, caused by obstruction to the outflow of urine. The
obstruction may be sudden or insidious, and it may occur at any level of the urinary
tract, from the urethra to the renal pelvis. The most common causes are
1. Congenital: e.g. urethral atresia, ureteric or urethral valves, aberrant renal artery
2. Acquired:
a. Foreign bodies: calculi
b. Tumors of the prostate or bladder (benign or malignant)
c. Contiguous malignant disease: (retroperitoneal lymphoma, carcinoma of the cervix
or uterus)
d. Inflammation: prostatitis, ureteritis, urethritis
e. Neurogenic: Spinal cord damage with paralysis of the bladder
Oxalate stone
Uric acid stone
staghorn phosphate stone

f. Normal pregnancy: mild and reversible
Bilateral hydronephrosis occurs only when the obstruction is below the level of the
ureters. If blockage is at the ureters or above, the lesion is unilateral. Sometimes
obstruction is complete, allowing no urine to pass; usually it is only partial.
Pathogenesis
Even with complete obstruction, glomerular filtration persists for some time, and the
filtrate subsequently diffuses back into the lymphatic and venous systems. Because of
the continued filtration, the affected calyces and pelvis become dilated, often
markedly so. The unusually high pressure thus generated in the renal pelvis, as well as
that transmitted back through the collecting ducts, causes compression of the renal
blood vessels. Both arterial insufficiency and venous stasis result. The most severe
effects are seen in the papillae, because they are subjected to the greatest increases in
pressure. Accordingly, the initial functional disturbances are largely tubular,
manifested primarily by impaired concentrating ability. Thereafter the glomerular
filtration begins to diminish. Serious irreversible damage occurs in about 3 weeks
with complete obstruction, and in 3 months with incomplete obstruction.
Gross features
The changes vary with the degree and speed of obstruction
- With subtotal or intermittent obstruction, the kidney is massively enlarged
consisting almost entirely of the greatly distended pelvicalyceal system. The
renal parenchyma shows compression atrophy, with obliteration of the papillae
and flattening of the pyramids.
- With sudden complete obstruction, glomerular filtration is reduced relatively
early, and as a consequence, renal function may cease while dilation is still
slight.
Depending on the level of the obstruction, one or both ureters may also be dilated
(hydroureter).
Hydronephrosis:
There is marked
dilation of the
pelvi-calyceal
system and
thinning of renal
parenchyma.

Microscpic features
Early lesions show tubular dilation, followed by atrophy and fibrous replacement
with relative sparing of the glomeruli. Eventually, in severe cases the glomeruli also
become atrophic and disappear, converting the entire kidney into a thin shell of
fibrous tissue.
With sudden and complete obstruction, there may be coagulative necrosis of the
renal papillae.
Course & prognosis
Bilateral complete obstruction produces anuria. When the obstruction is below the
bladder, there is bladder distention. Incomplete bilateral obstruction causes polyuria
rather than oliguria, as a result of defects in tubular concentrating mechanisms.
Unilateral hydronephrosis may remain completely silent. Removal of obstruction
within a few weeks usually permits full return of function; however, with time the
changes become irreversible.
TUMORS
The kidney is the site of benign and malignant tumors. In general, benign tumors
(such as small cortical adenomas or medullary fibromas) have no clinical significance.
The most common malignant tumors of the kidney in descending order of frequency
are
1. Renal cell carcinoma
2. Nephroblastoma (Wilms tumor)
3. Transitional cell carcinoma of the calyces and pelvis
Renal Cell Carcinoma (RCC)
This is derived from the renal tubular epithelium and represents the most common
primary malignant tumor of the kidney (80%). The mean age of incidence is 50 to 70
years. Risk factors include:
smoking,
hypertension,
obesity,
occupational exposure to cadmium,
and acquired polycystic disease complicating chronic dialysis (30-fold).
RCC are currently classified according to the molecular origins of these tumors. The
three most common forms are
1. Clear Cell Carcinomas (80% of RCCs): these are made up of cells with clear or
granular cytoplasm. The majority are sporadic, but may occur in familial forms or in
association with von Hippel-Lindau disease (VHLD). The latter is autosomal
dominant and characterized by predisposition to a variety of neoplasms including
bilateral & often multiple RCC of clear cell type. Patients with VHLD inherit a germ-
line mutation of the VHL gene on chromosome 3 and lose the second allele by
somatic mutation. Thus, the loss of the normal copies of both these tumor suppressor
genes gives rise to clear cell carcinoma. It has been also confirmed that homozygous
loss of the VHL gene seems to be the common underlying molecular abnormality in
both sporadic and familial forms of clear cell carcinomas.
2. Papillary Renal Cell Carcinomas (10%). These tumors are frequently multifocal
and bilateral and like clear cell carcinomas, they occur in familial and sporadic forms.
The cause is the MET proto-oncogene. It is the increased dosage of the MET gene

(due to duplications of chromosome 7) seems to encourage neoplastic growth
abnormal growth in the proximal tubular epithelial cell precursors.
3. Chromophobe RCCs: these are the least common; they arise from intercalated cells
of collecting ducts. Their name indicates that the tumor cells stain more darkly than
cells in clear cell carcinomas. These tumors are unique in having multiple losses of
entire chromosomes, including chromosomes 1 and 2. In general, they have a good
prognosis.
Pathologic features
Clear cell carcinoma
Gross features:
There is usually solitary spherical mass; large when symptomatic.
The cut surface is variegated; yellow-orange to gray-white with red areas of
hemorrhage. There are prominent areas of cystic degeneration.
Although the margins of the tumor are well defined, with enlargement extension
may occur in two directions
1. Into the pelvicalyceal system & as far down as the ureter.
2. Into the renal vein, then the inferior vena cava & even into the right side of
the heart.
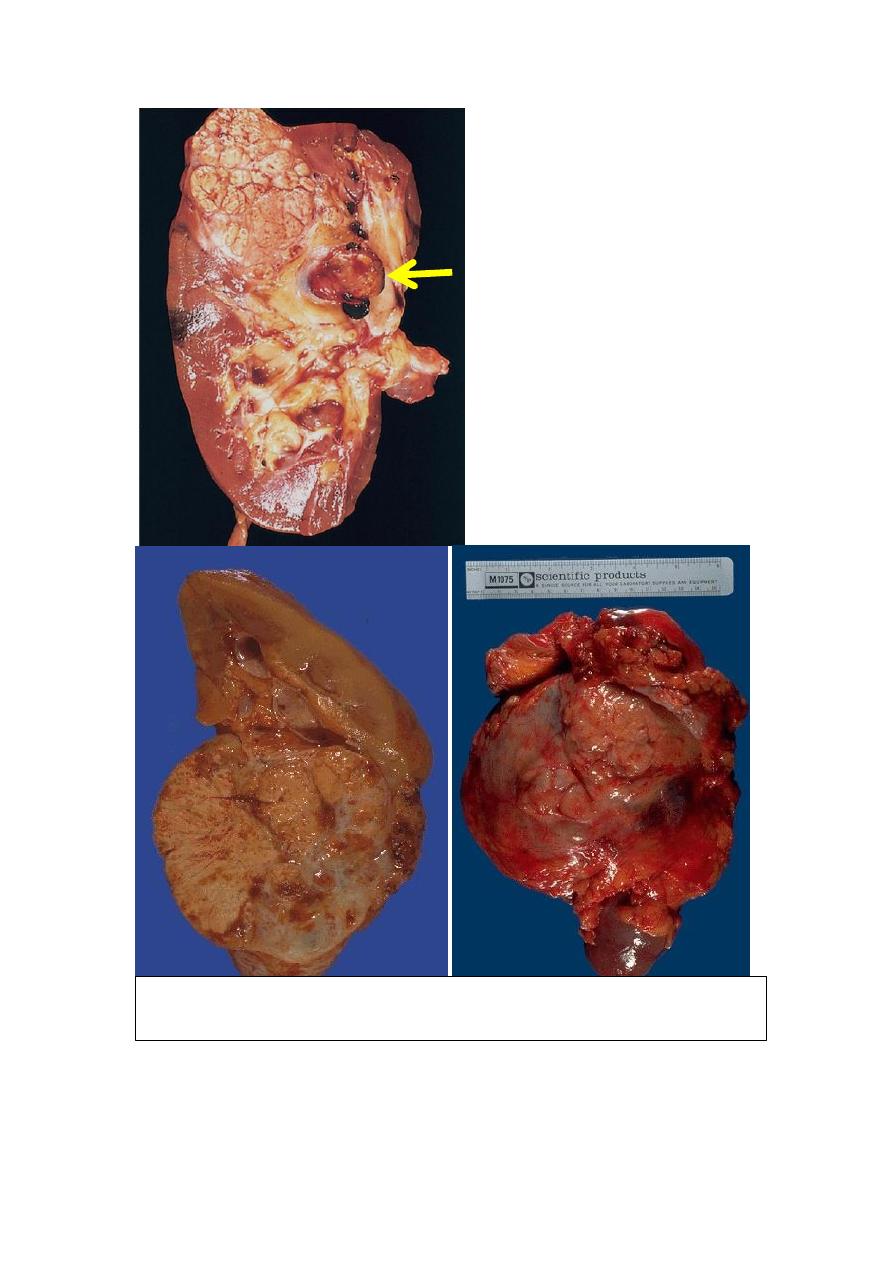
Microscopic features
The classic lipid- & glycogen-laden clear cells have well defined cell membranes.
The nuclei are usually small and round.
Typical cross-section of
yellowish, spherical neoplasm
in one pole of the kidney. Note
the tumor in the dilated,
thrombosed renal vein (arrow).
The varrigated appearance of RCC (yellowish-brownish with areas of
hemorrhage and necrosis
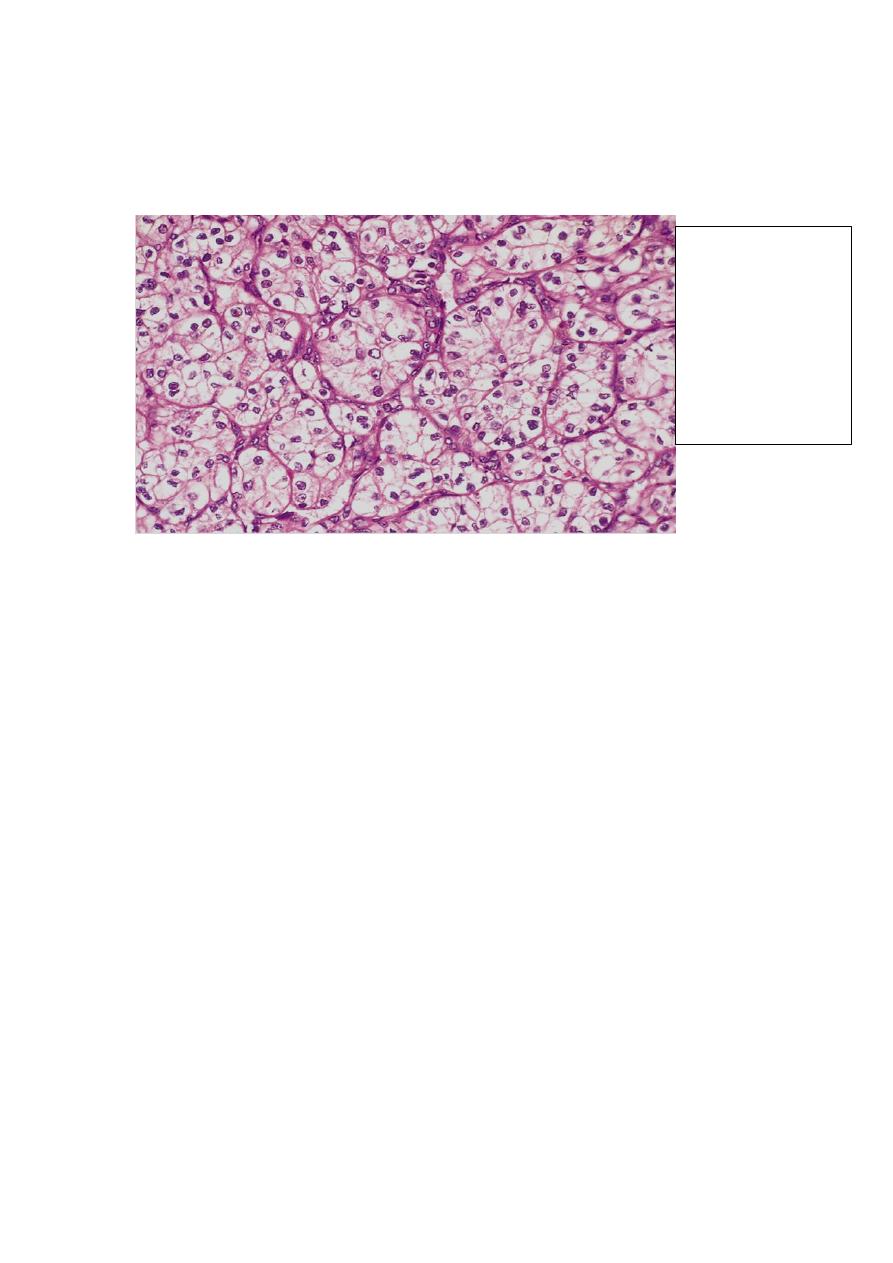
These cells are mixed to varying extent with cells having granular pink cytoplasm.
Some tumors exhibit marked degrees of anaplasia, with numerous mitotic figures
and markedly enlarged, hyperchromatic, pleomorphic nuclei (sarcomatoid RCC).
The stroma is usually scant but highly vascularized.
Papillary renal cell carcinomas
Exhibit papilla formation with fibrovascular cores.
They tend to be bilateral and multiple.
The cells can have clear or, more commonly, pink cytoplasm.
The classical triad of painless hematuria (the most frequent), a palpable abdominal
mass, and dull flank pain is characteristic. RCCs are well-known for their association
with several paraneoplastic syndromes. Polycythemia may occur; it results from
excessive secretion of erythropoietin by the tumor. Uncommonly, these tumors also
produce a variety of hormone-like substances, resulting in hypercalcemia,
hypertension, Cushing syndrome, etc. In many individuals the primary tumor remains
silent and is discovered only after its metastases have produced symptoms. Common
locations for metastases are the lungs and the bones. Forty percent of patients die of
the disease.
Wilms Tumor (WT) (nephroblastoma)
This is the most common primary renal tumor of children and the third most common
organ cancer in those younger than 10 years. WT may arise sporadically or be
familial. Some congenital malformations are associated with an increased risk of
developing Wilms' tumor such as aniridia, mental retardation, gonadal dysgenesis and
renal abnormalities. These conditions are associated with inactivation of the Wilms'
tumor 1 (WT1) gene, located on chromosome11. This gene is critical to normal renal
and gonadal development. Patients with the Beckwith syndrome (BWS);
characterized by enlargement of individual body organs (e.g., tongue, kidneys, or
liver) or hemihypertrophy of the entire body segments. In addition to Wilms' tumors,
patients with BWS are also at increased risk for developing other cancers e.g.
hepatoblastoma.
Well-defined nests
of the tumor cells
that appear clear
(lipid-laden) with
well defined cell
membranes. The
nuclei are usually
small and round
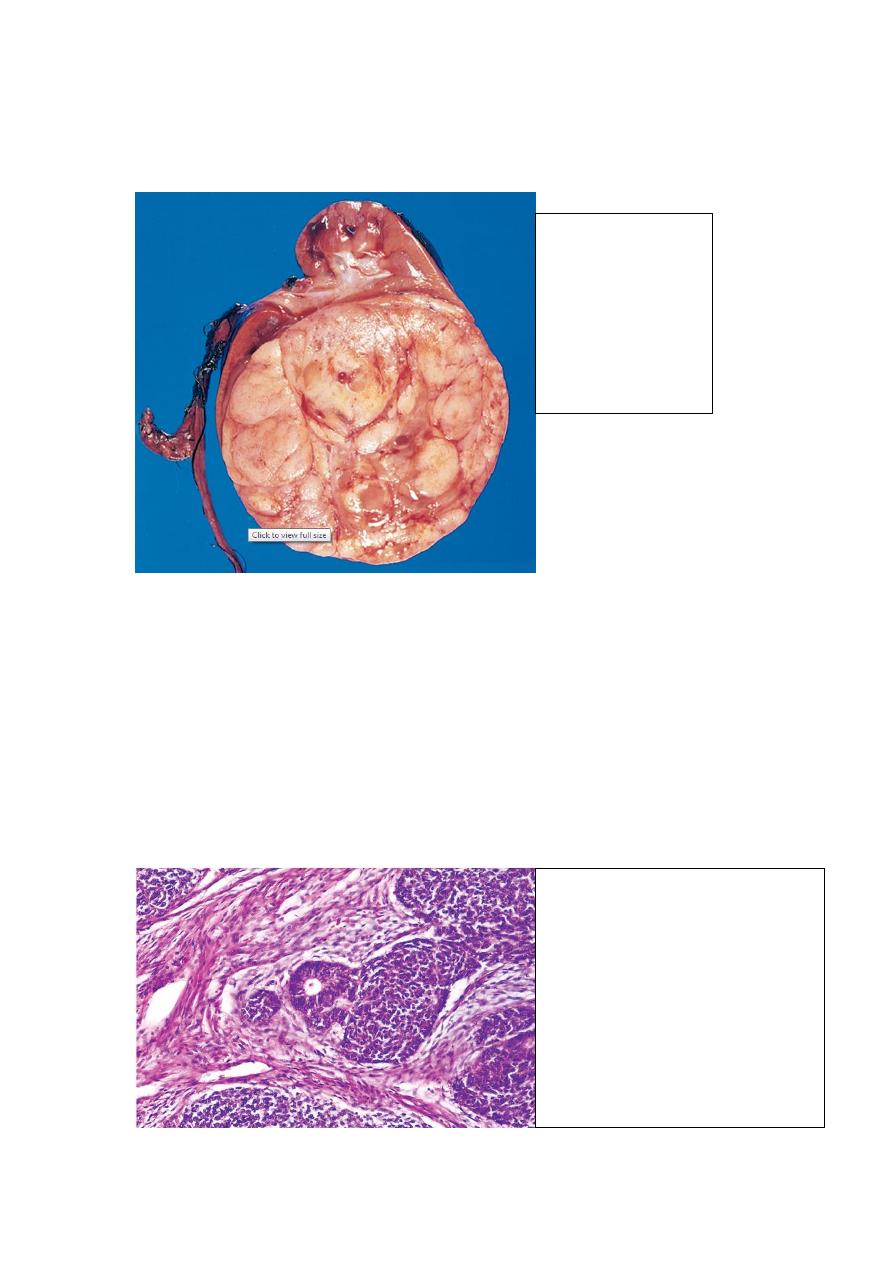
Gross features
WT tends to be large, well-circumscribed mass with soft, homogenous, tan to gray
cut section accentuated with occasional foci of hemorrhage, necrosis and cystic
degeneration.
Microscopic features
In WT there are attempts to recapitulate different stages of nephrogenesis.
WT contain a variety of tissue components, but all derived from the mesoderm.
The classic triphasic combination of blastemal, stromal, and epithelial cell types is
observed in most lesions.
- The blastemal component is represented by sheets of small blue cells
- The epithelial component (differentiation) is represented by abortive tubules
or glomeruli.
- The stromal component is represented usually by fibroblastic cells or
myxoid areas, although
skeletal
muscle
"differentiation"
and
other
mesenchymal elements may be seen.
The presence of anaplasia correlates with underlying p53 mutations, and the
emergence of resistance to chemotherapy.
Wilm's tumor:
The tumor is in the
lower pole of the
kidney with the
characteristic tan to
gray color and well-
circumscribed
margins.
Triphasic histology of Wilms'
tumor: the stromal component is
composed of spindle-shaped cells
in the less cellular area on the left;
the immature tubule in the center
is an example of the epithelial
component, and the tightly packed
blue cells the blastemal elements.

Clinically, there is a readily palpable abdominal mass, which may extend across the
midline and down into the pelvis. Fever and abdominal pain, with hematuria, are less
frequent. The prognosis of Wilms' tumor is generally very good, and excellent results
are obtained with a combination of nephrectomy and chemotherapy. WTs with diffuse
anaplasia, especially those with extra-renal spread, have the least favorable outcome.
RENAL PELVIS, URETER, URINARY BLADDER & URTHERA
URETERS
Congenital Anomalies these are rare, and mostly of little clinical significance.
However, some may contribute to urine outflow obstruction. Incompetent
ureterovesical junction predisposes to pyelonephritis. The majority of double ureters
are unilateral and of no clinical significance. A congenital ureteropelvic junction
obstruction is the most common cause of hydronephrosis in infants and children.
There is agenesis of the kidney on the opposite side in a significant number of cases
.
Diverticula are saccular outpouchings acting as pockets of stasis and secondary
infections. Hydroureter with elongation and tortuosity may be congenital leading to
hydronephrosis if untreated.
Ureteritis may be one component of UTI. With long-standing chronic ureteritis, there
may be aggregation of lymphocytes in the subepithelial region causing fine granular
mucosal surface (ureteritis follicularis), or the mucosa may become sprinkled with
tiny cysts (ureteritis cystica). Identical changes are found in the bladder. The two most
common tumors and tumor-like lesions
are fibroepithelial polyps and leiomyomas.
Primary malignant tumors are similar to those arising in the renal pelvis, calyces, and
bladder; the majority are transitional cell carcinomas. They cause obstruction and are
most frequently during the sixth and seventh decades. They can be multiple and may
occur concurrently with similar tumors in the bladder or renal pelvis.
Obstructive lesions of the ureters
A great variety of pathologic lesions may obstruct the ureters and give rise to
hydroureter, hydronephrosis, and sometimes pyelonephritis. The more important
causes include
1. impacted small stones
2. strictures (congenital or acquired),
3. primary carcinoma,
4. pregnancy,
5. retroperitoneal fibrosis & cancers of the rectum, bladder, prostate, ovaries, uterus
and cervix.
Sclerosing Retroperitoneal Fibrosis is a fibrous proliferative inflammatory process
encasing the retroperitoneal structures including the ureters and causing
hydronephrosis. Two thirds of the cases are idiopathic. The remaining cases are
attributed to drugs (ergot derivatives, β-adrenergic blockers), adjacent inflammatory
conditions or malignant disease. It may be associated with mediastinal fibrosis,
sclerosing cholangitis, and Riedel thyroiditis. This suggests that the disorder is
systemic in distribution but preferentially involves the retroperitoneum. An
autoimmune reaction, sometimes triggered by drugs, has been proposed.
URINARY BLADDER
Congenital anomalies
Diverticula are pouch-like evaginations of the bladder wall that may be congenital,
but more commonly acquired due to persistent urethral obstruction (e.g. prostatic

hyperplasia or neoplasia). In both forms, there are frequently multiple sac-like
pouches that range from less than 1 cm to 10 cm in diameter. Most diverticula are
small and asymptomatic, but may be sites of urinary stasis that predispose to infection
and the formation of bladder calculi.
Exstrophy is a developmental failure in the anterior wall of the abdomen and the
bladder, so that the bladder communicates directly through a large defect with the
surface of the body. The exposed bladder mucosa may undergo colonic glandular
metaplasia and is subjected to infections. There is an increased risk of carcinoma.
Vesicoureteral reflux is the most common and serious anomaly that contributes to
renal infection and scarring.
Congenital fistulas are abnormal connections between the bladder and the vagina,
rectum, or uterus.
Persistent urachus refers to failure of the urachus to close in part or in whole. When
it is totally patent, a fistulous urinary tract is created that connects the bladder with the
umbilicus. Sometimes, only the central region of the urachus persists, giving rise to
urachal cysts. Carcinomas, mostly adenocarcinomas, may arise in such cysts.
ACUTE & CHRONIC CYSTITIS
The common etiologic agents of bacterial cystitis are the E. coli, followed by Proteus,
&
Klebsiella. Women are more likely to develop cystitis due to their shorter urethras.
Bacterial pyelonephritis is frequently preceded by cystitis, with retrograde spread of
microorganisms into the kidneys and their collecting systems. Tuberculous cystitis is
almost always a consequence of renal tuberculosis. Fungal cystitis is usually due to
Candida albican. It is particularly seen in immunosuppressed patients or those
receiving long-term antibiotics. Schistosomal cystitis (Schistosoma haematobium) is
common in certain Middle Eastern countries, notably Egypt. Viruses (e.g.,
adenovirus), Chlamydia, and Mycoplasma may also be causes of cystitis.
Predisposing factors of cystitis include
1. Urinary obstruction e.g. prostatic hyperplasia, bladder calculi,tumors
2. cystocele or diverticula
3. Diabetes mellitus
4. Instrumentation
5. Immune deficiency.
Hemorrhagic cystitis sometimes complicates cytotoxic antitumor drugs (e.g.
cyclophosphamide). Radiation cystitis is due to radiation of the bladder region.
Most cases of cystitis take the form of nonspecific acute or chronic inflammation of
the bladder. Gross features
There is hyperemia of the mucosa.
Hemorrhagic cystitis shows in addition a hemorrhagic component; this form is
sometimes follows radiation injury, antitumor chemotherapy, or adenovirus infection.
Suppurative cystitis is characterized by the accumulation of large amounts of
suppurative exudate.
Ulcerative cystitis refers to cystitis associated with ulceration of large areas of the
mucosa, or sometimes the entire bladder mucosa.
Persistence of the infection leads to chronic cystitis, which shows red, friable,
granular, sometimes ulcerated mucosa. Chronicity is also associated with fibrous
thickening and inelasticity of the bladder wall.
Microscopic features
In acute cystitis there are the expected features of acute inflammation.
In chronic forms there is chronic inflammatory cells infiltration with fibrosis.
Variants of chronic cystitis include Follicular cystitis and Eosinophilic cystitis
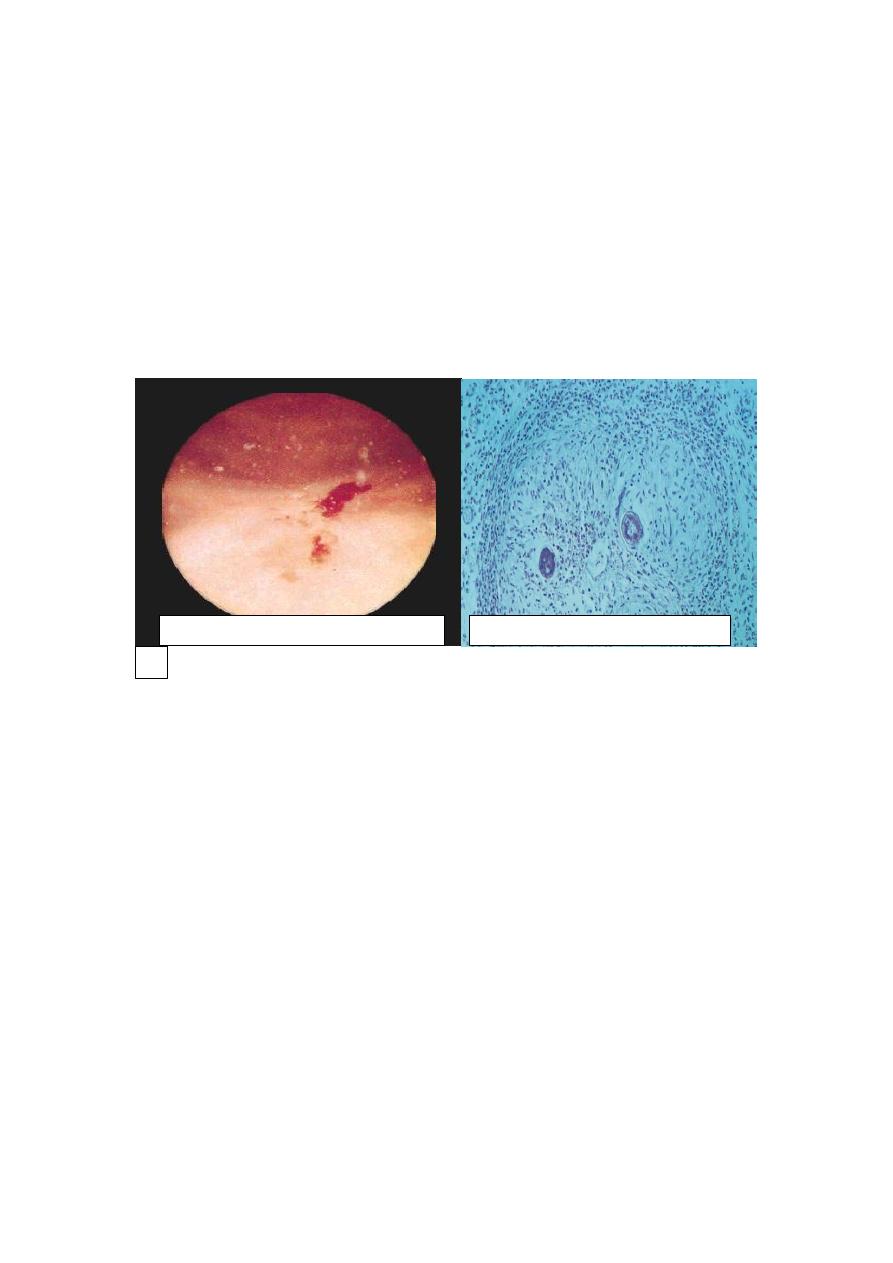
Schistomal cystitis: urogenital bilharziasis is caused by S. haematobium. Eggs are
deposited in the superior rectal vein. From there, they pass through anastomoses into
the veins of the wall of urinary bladder. There they cause granulomatous cystitis with
eosinophilic infiltrate & fibrosis. These granulomas are visible under endoscopy as
minute granules referred to as “sand grain” cystitis. The eggs eventually die in the
tissue with regressive calcification.The condition may be complicated by
a. Extensive fibrosis that may impinge on the ureteric orifices with eventual
hydronephrosis
b. Carcinoma of bladder that is frequently squamous in type; as this form of cystitis
can be associated with squamous metaplasia of the native transitional epithelium.
Special Forms of Cystitis
These are distinctive by either their morphologic appearance or their causation.
1. Interstitial Cystitis (Hunner Ulcer): a painful form of chronic cystitis occurring
most frequently in women. Cystoscopy shows fissures and punctate hemorrhages in
the mucosa, sometimes with chronic mucosal ulcers (Hunner ulcers). Infiltration by
mast cells is characteristic of this disease. The condition may be of autoimmune
origin.
2. Malakoplakia is characterized macroscopically by soft, yellow, slightly raised
mucosal plaques 3 to 4 cm in diameter and histologically by infiltration with large,
foamy macrophages with debris of bacterial origin (mostly E. coli) (Fig. 21-27). In
addition, laminated mineralized concretions (Michaelis-Gutmann bodies) are typically
present. Similar lesions have been described in other organs e.g. colon, lungs, bones.
It occurs with increased frequency in immuno-suppressed transplant recipients and as
a result of defects in phagocytic or degradative function of macrophages.
3. Polypoid Cystitis is an inflammatory condition resulting from irritation to the
bladder mucosa mostly by indwelling catheters. The urothelium is thrown into broad,
bulbous, polypoid projections as a result of marked submucosal edema.
METAPLASTIC LESIONS
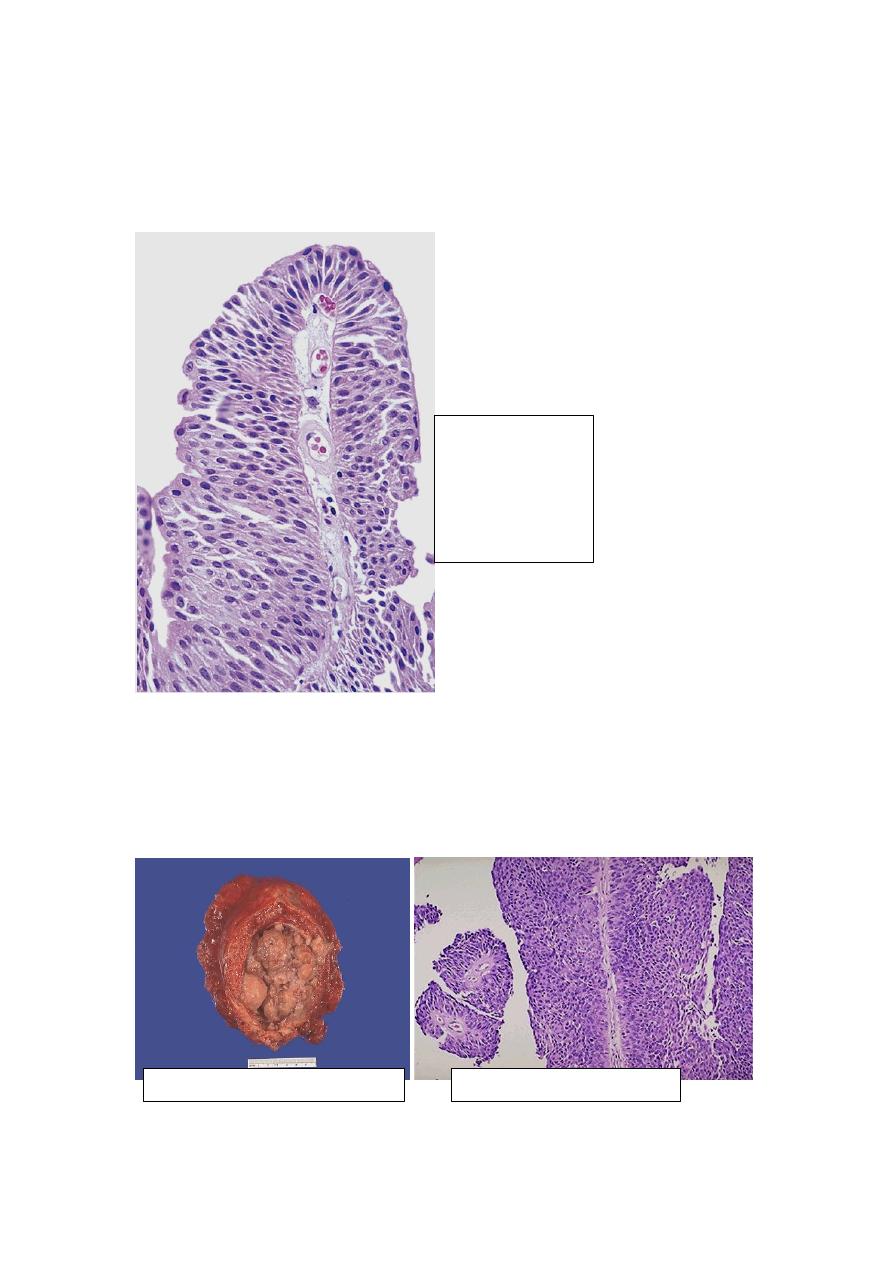
Cystitis Glandularis and Cystitis Cystica: these terms refer to common lesions in
which nests of transitional epithelium (Brunn nests) grow downward into the lamina
propria and undergo transformation of their central epithelial cells into columnar
epithelium lining (cystitis glandularis) or cystic spaces lined by urothelium (cystitis
Bilharzial (“sand grain”) cystitis
Schistosomic granuloma (HE)
x 150
cystica). The two processes often coexist. In a variant of cystitis glandularis, goblet
cells are present (intestinal metaplasia).
Both variants are common microscopic incidental findings in relatively normal
bladders and are not associated with an increased risk of adenocarcinoma.
Two forms of metaplasia occur in response to injury
1. Squamous Metaplasia
2. Nephrogenic Metaplasia (Nephrogenic Adenoma): the urothelium may be focally
replaced by cuboidal epithelium, which can assume a papillary growth pattern with
subjacent tubular proliferation.
TUMORS OF THE URINARY BLADDER AND COLLECTING SYSTEM
(Renal Calyces, Renal Pelvis, Ureter, and Urethra)
The entire urinary collecting system from renal pelvis to urethra is lined with
transitional epithelium, so its epithelial tumors assume similar morphologic patterns.
Tumors in the collecting system above the bladder are relatively uncommon; those in
the bladder, however, are a more frequent cause of death than are kidney tumors.
Gross features
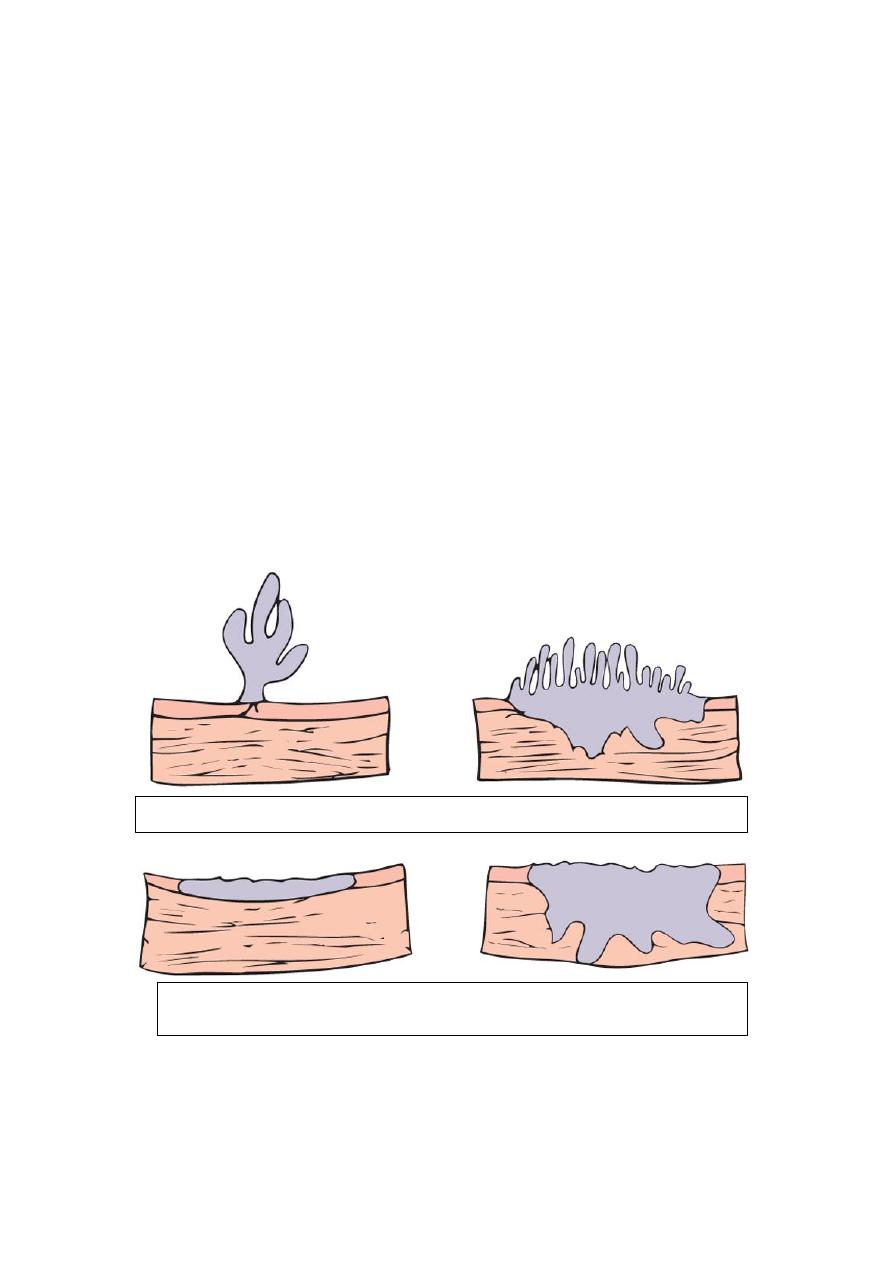
Four morphologic patterns are recognized that range from small benign papillomas to
large invasive cancers. These tumors are classified into
1. Benign papilloma (rare)
2. Papillary urothelial neoplasms of low malignant potential, and
3. Urothelial carcinoma (low and high grade)
Papillomas are very rare, small (up to 1 cm) benign tumors with frondlike structures
having a delicate fibrovascular core covered by multilayered, well-differentiated
Papilloma-papillary carcinoma
Invasive papillary
carcinoma
Flat non-invasive carcinoma
Flat invasive carcinoma
transitional epithelium. Such lesions are usually solitary. They rarely recur once
removed.
Urothelial (transitional) cell carcinomas range from papillary to flat, noninvasive to
invasive and low grade to high grade.
Low-grade carcinomas are always papillary and are rarely invasive, but they may
recur after removal.
High-grade carcinoma can be papillary or occasionally flat; they may cover larger
areas of the mucosal surface, invade deeper, and have a shaggier necrotic surface than
do low-grade tumors. Occasionally, these cancers show foci of squamous cell
differentiation, but only 5% of bladder cancers are true squamous cell carcinomas.
Carcinomas of grades II and III infiltrate surrounding structures, spread to regional
nodes, and, on occasion, metastasize widely.
Low-grade
papillary
urothelial
carcinoma of the
bladder
Exophytic TCC of urinary
bladder
High grade Papillary TCC
Painless hematuria is the dominant clinical presentation of all these tumors. They
affect men three times more frequently than women and usually develop between
the ages of 50 and 70 years.
Risk factors of bladder cancer are
1. Exposure to β-naphthylamine (50 times increased risk).
2. Cigarette smoking
3. Chronic cystitis
4. schistosomiasis of the bladder
5. Certain drugs (cyclophosphamide).
A wide variety of genetic abnormalities are seen in bladder cancers; of these,
mutations involving several genes on chromosome 9, p53, and FGFR3 are the most
common.
The prognosis of bladder tumors depends on their histologic grade and the depth of
invasion of the lesion; the latter is much more important. Except for benign
papillomas, all tend to recur after removal. Lesions that invade the ureteral or urethral
orifices cause urinary tract obstruction. Overall 5-year survival is 57%. With deep
penetration of the bladder wall the 5-year survival rate is less than 20%.
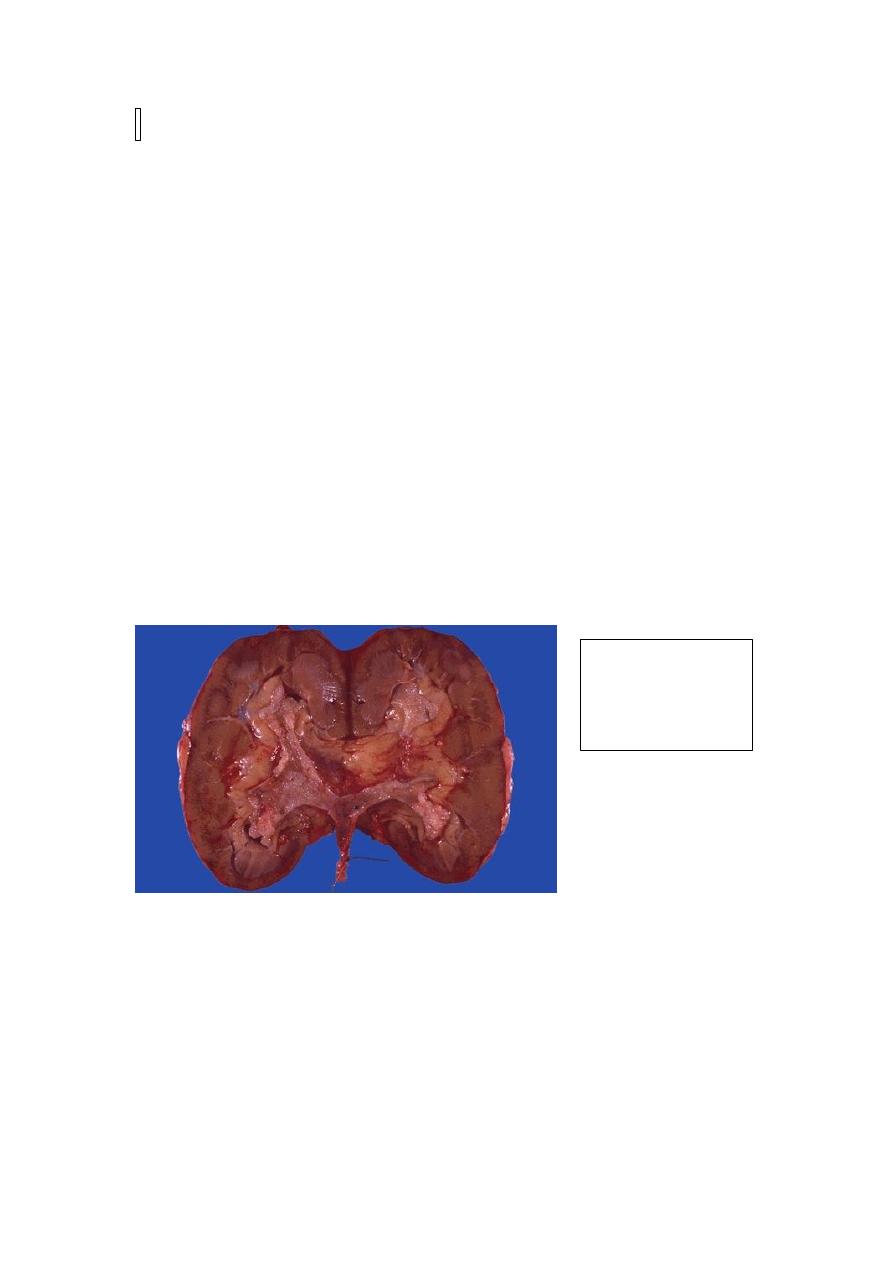
Papillary tumors occur much less frequently in the renal pelvis than in the bladder,
they nonetheless make up to 10% of primary renal tumors. Patients present with
painless hematuria, and may develop hydronephrosis. Infiltration of the walls of the
pelvis, calyces, and renal vein worsens the prognosis. Despite removal of the tumor
by nephrectomy, fewer than 50% of patients survive for 5 years.
Cancer of the ureter is fortunately the rarest of the tumors of the collecting system.
The 5-year survival rate is less than 10%.
Papillary TCC of
renal pelvis