
Bleeding Disorders:
(Hemorrhagic Diatheses)

• The normal haemostatic response to vascular
damage depends on a closely linked
interaction between the:
• 1-blood vessel wall
• 2-Circulating platelets
• 3-Blood coagulation factors

• Excessive bleeding can result from:
• 1. Increased fragility of vessels.
• 2. Platelet deficiency or dysfunction.
• 3. Derangement of coagulation.
• 4. Combinations of these.

Tests used to evaluate different
aspects of hemostasis are the
following:

1. Bleeding time:
2. Platelets count:
3. Prothrombin time (PT):
4. Partial thromboplastin time (PTT):
5. Other tests

1-Bleeding time:
• This measures the time taken for a
standardized skin puncture to stop bleeding
and provides an in vivo assessment of platelet
response to limited vascular injury.

Bleeding time:
cont.
The reference range depends on the actual
method employed and varies from
2
to
9
minutes.
•
Prolongation generally indicates a defect in
platelet numbers or function.

2-Platelets count:
•
These are obtained on anticoagulated blood
using an electronic particle counter.
•
The reference range is 150 to 400 × 103/μL.

3-Prothrombin time (PT):
•
This assay tests the extrinsic and common
coagulation pathways.
•
The normal time for clotting is
10-14 s.
•
•
A prolonged PT can result from deficiency or
dysfunction of: factor VII, factors X, V,
prothrombin, or fibrinogen.

4-Partial thromboplastin time (PTT):
•
This assay tests the intrinsic and common clotting
pathways.
•
The normal time for clotting is approximately
30-
40s
.
•
•
Prolongation of the PTT can be due to deficiency
or dysfunction of: factors VIII, IX, XI, or XII, factors
X, V, prothrombin, or fibrinogen.

Q.1-A 25-year-old man has a lifelong
hemorrhagic diathesis. The
Prothrombin
time (PT) and bleeding time are
normal
, but
the
Partial thromboplastin time (PTT)
is
prolonged
. The most likely cause of the
bleeding disorder is:
a. Factor VIII deficiency.
b. Factor X deficiency.
c. Factor VII deficiency.
d. A platelet functional disorder.
e. von Willebrand disease.

Q.1-A 25-year-old man has a lifelong
hemorrhagic diathesis. The
Prothrombin
time (PT) and bleeding time are
normal
, but
the
Partial thromboplastin time (PTT)
is
prolonged
. The most likely cause of the
bleeding disorder is:
a. Factor VIII deficiency.
b. Factor X deficiency.
c. Factor VII deficiency.
d. A platelet functional disorder.
e. von Willebrand disease.

Bleeding Disorders Caused By Vessel Wall
Abnormalities:
•
Disorders within this category, sometimes
called nonthrombocytopenic purpuras, are
•
relatively common but do not usually cause
serious bleeding problems.

Bleeding Disorders Caused By Vessel Wall
Abnormalities:
cont.
•
Most often, they induce small hemorrhages
(petechiae and purpura) in the skin or mucous
membranes, particularly the gingiva.
•
نمشات
Petechiae =
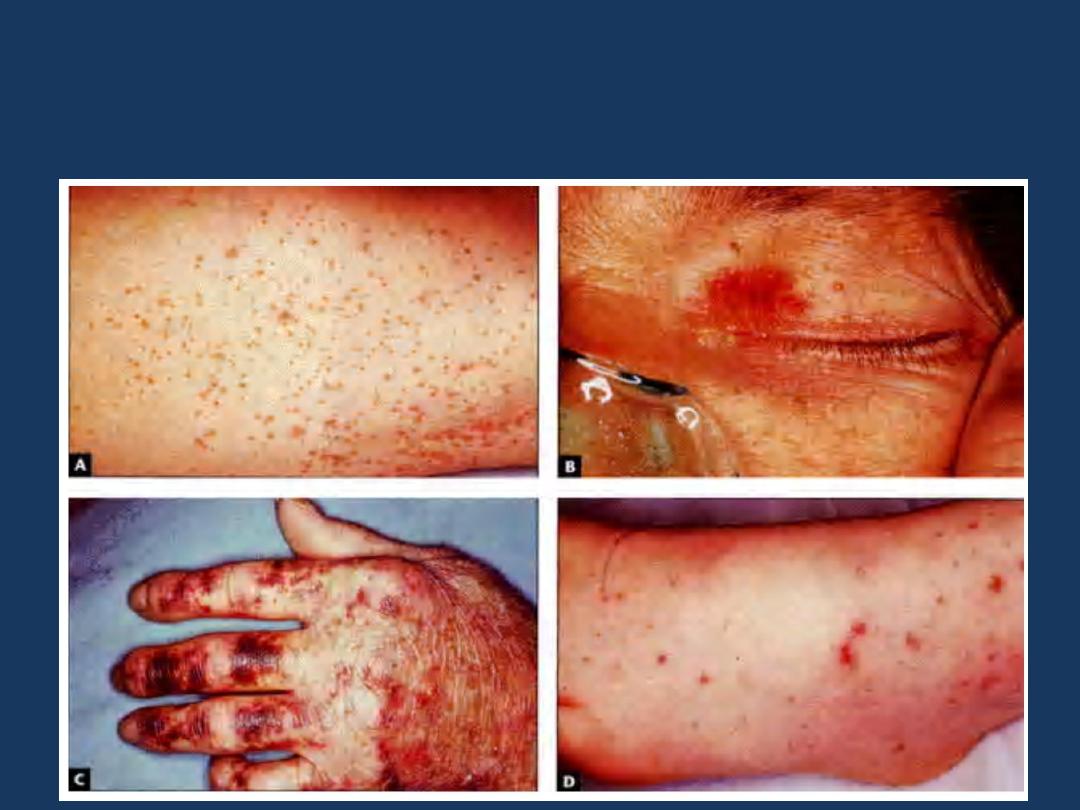
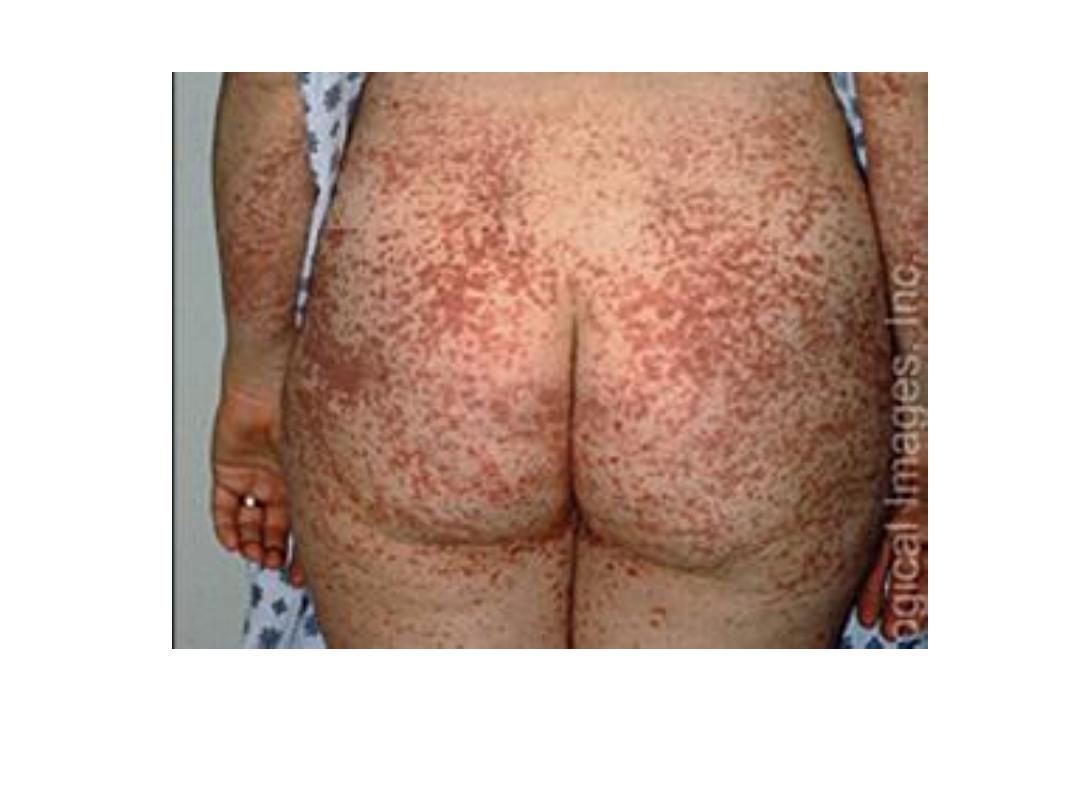
BLEEDING DISORDERS CAUSED BY VESSEL WALL
ABNORMALITIES
They induce small hemorrhages (petechiae and purpura) in the skin or
mucous membranes, particularly the gingivae.
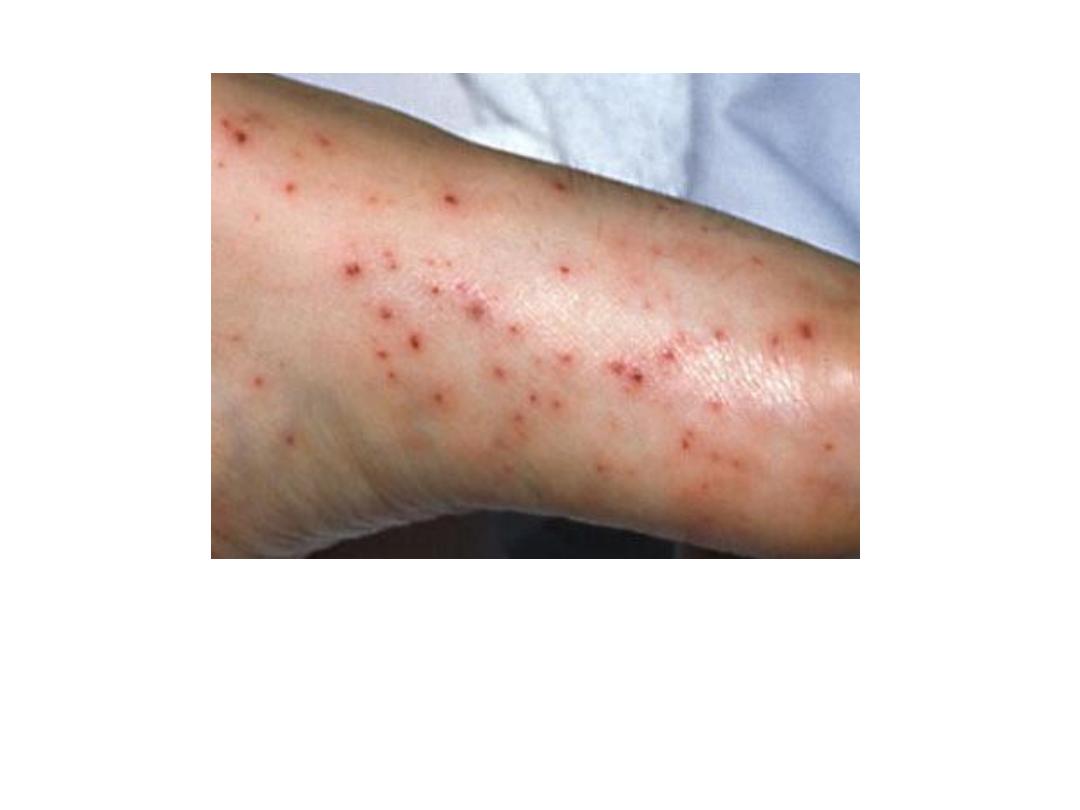
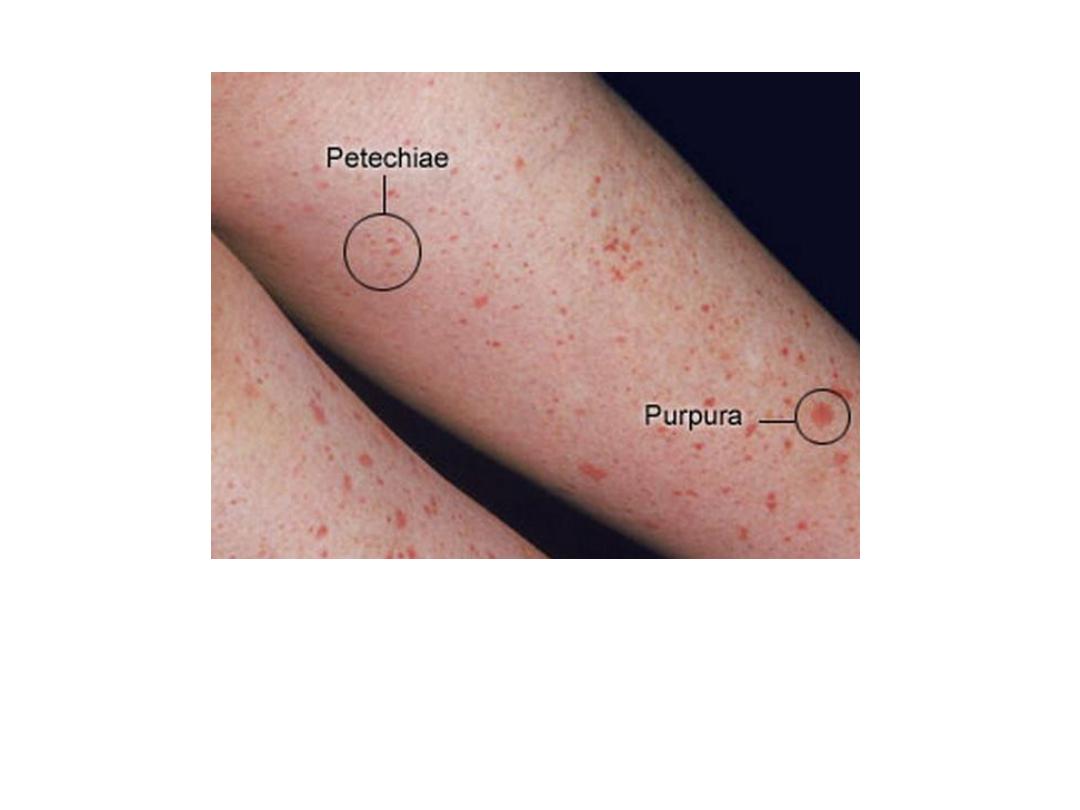
Petechiae
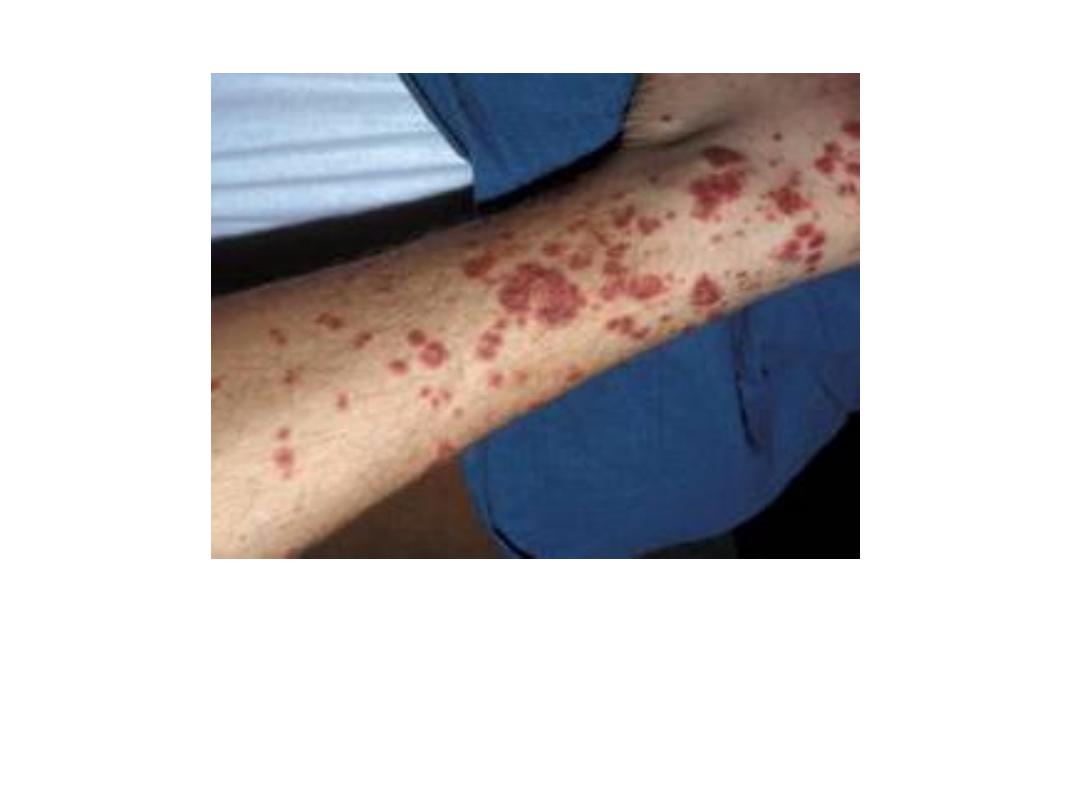
Purpura

Bleeding Disorders Caused By Vessel Wall
Abnormalities:
cont.
•
The platelet count, bleeding time, and results
of the coagulation tests (PT, PTT) are usually
normal.

•
The varied clinical conditions in
which hemorrhages can be related to
abnormalities in the vessel wall
include the following:

•
•Many
infections
induce petechial and
purpuric hemorrhages, but especially
implicated are:
•
1- meningococcemia,
•
2- other forms of septicemia,
•
3- infective endocarditis,
•
4- and several of the rickettsioses.
•
•
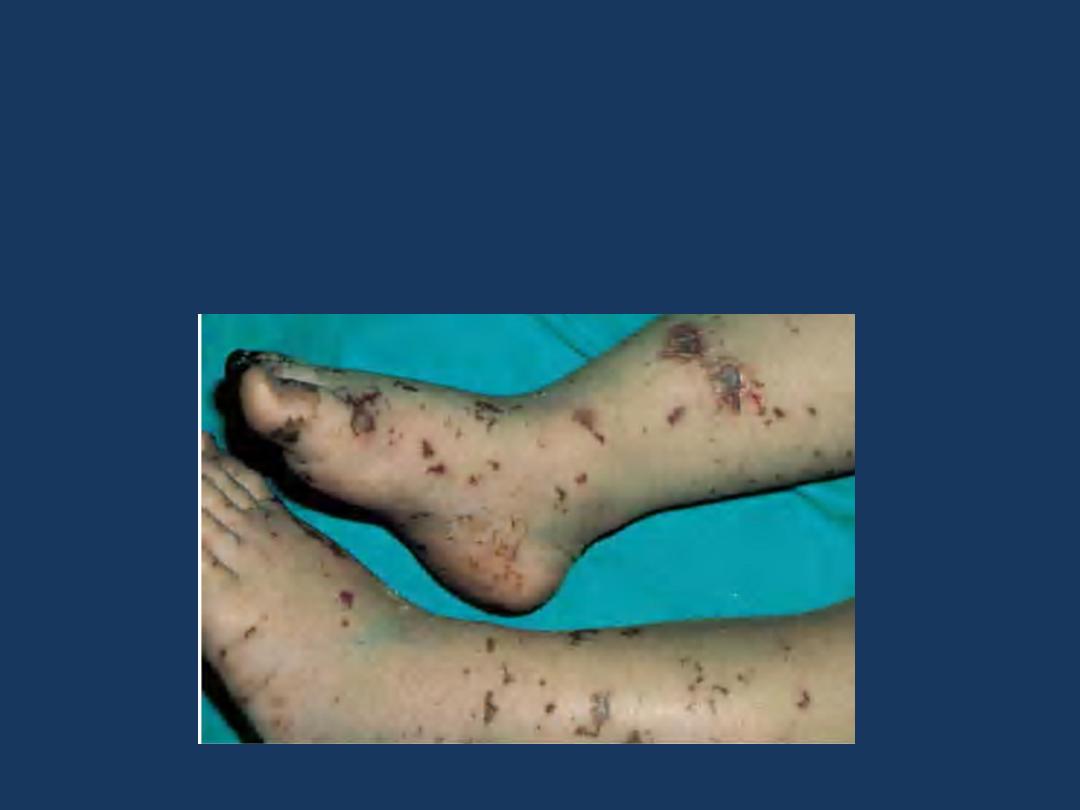
Clinical
conditions
in
which
hemorrhages
can
be
related
to
abnormalities in the vessel wall include the following:
•
Many
infections:
induce petechial and purpuric hemorrhages.
The involved
mechanism
is presumably microbial damage to the
microvasculature
(vasculitis)
or disseminated intravascular coagulation
(DIC).
Meningococcemia: stellate purpura.

•
The involved mechanism is presumably
microbial damage to the microvasculature
(vasculitis) or disseminated intravascular
coagulation (DIC).

•
Drug reactions
sometimes induce cutaneous
petechiae and purpura without causing
thrombocytopenia.
•
In many instances, the vascular injury is
mediated by drug-induced antibodies and
deposition of immune complexes in the vessel
walls, leading to hypersensitivity
(leukocytoclastic) vasculitis.
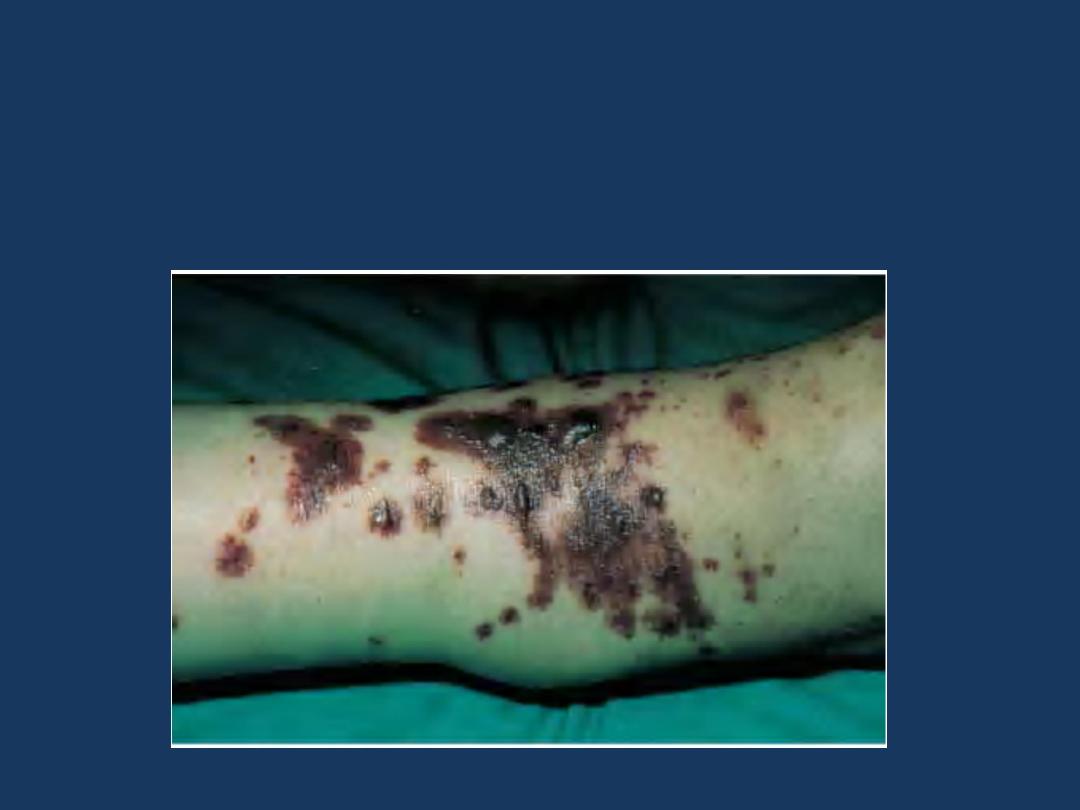
•
Drug reactions
sometimes induce cutaneous petechiae and
purpura without causing thrombocytopenia.
The involved
mechanism
is by drug-induced antibodies and
deposition of immune complexes in the vessel walls, leading to
hypersensitivity (leukocytoclastic) vasculitis
Leukocytoclastic vasculitis secondary to furosemide.

•
•
Scurvy, Cushing syndrome and Ehlers-
Danlos syndrome
are associated with
microvascular bleeding resulting from
impaired formation of collagens needed
for support of vessel walls.
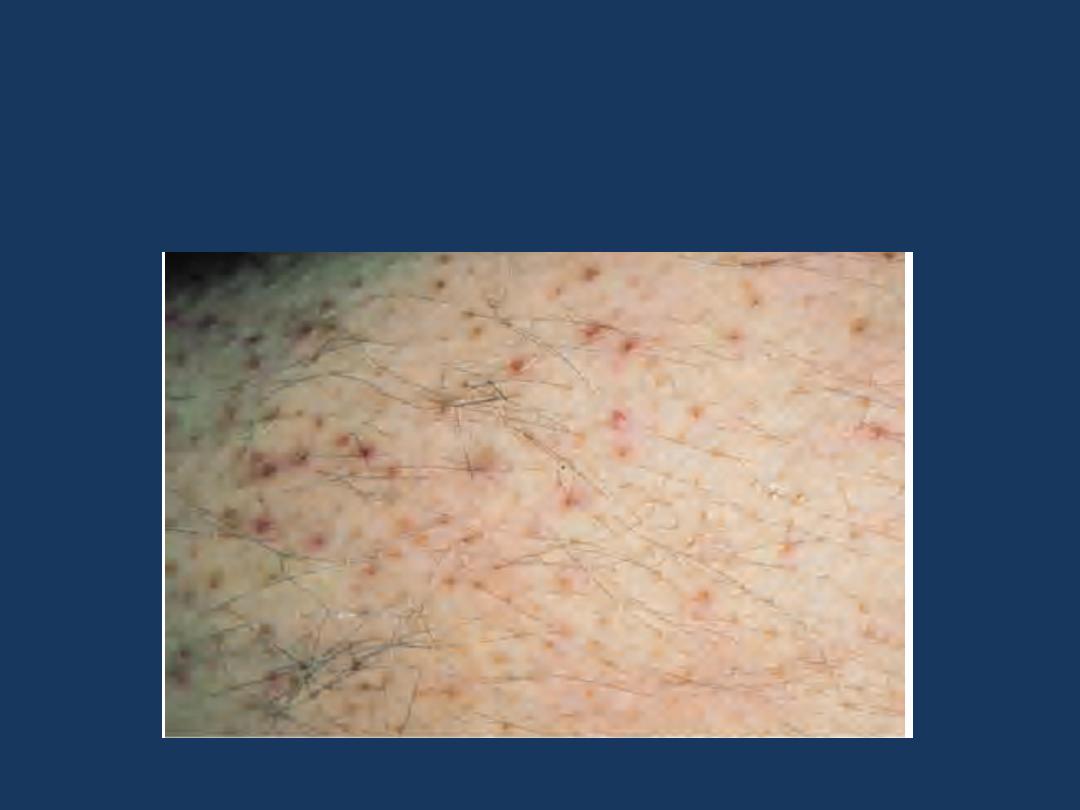
•
Scurvy and the Ehlers-Danlos syndrome
are associated with
microvascular bleeding.
The involved
mechanism
is
impaired formation of collagens
needed for support of vessel walls.
Scurvy: Vitamin C deficiency: Note parafollicular petechiae

Q.2-A 76-year-old female notices that small, pinpoint areas
of superficial hemorrhage have appeared on her gums and
on the skin of her arms and legs over several weeks. She is
found to have
a
normal
prothrombin time (PT)
and
partial
thromboplastin time (PTT)
. Her CBC shows a
hemoglobin
concentration of 12.7 g/dL, platelet count of 260,000/µL,
and WBC count of 8600/µL.
Her
template bleeding time is
3 minutes
.
Which o the following conditions best explains
these findings?
a. Macronodular cirrhosis.
b. Chronic renal failure.
c. Meningococcemia.
d. Metastatic carcinoma.
e. Vitamin C deficiency.

Q.2-A 76-year-old female notices that small, pinpoint areas
of superficial hemorrhage have appeared on her gums and
on the skin of her arms and legs over several weeks. She is
found to have
a
normal
prothrombin time (PT)
and
partial
thromboplastin time (PTT)
. Her CBC shows a
hemoglobin
concentration of 12.7 g/dL, platelet count of 260,000/µL,
and WBC count of 8600/µL.
Her
template bleeding time is
3 minutes
.
Which o the following conditions best explains
these findings?
a. Macronodular cirrhosis.
b. Chronic renal failure.
c. Meningococcemia.
d. Metastatic carcinoma.
e. Vitamin C deficiency.

•
•
Henoch-Schönlein purpura is a systemic
hypersensitivity disease of unknown
cause characterized by a purpuric rash,
colicky abdominal pain (presumably due
to focal hemorrhages into the
gastrointestinal tract), polyarthralgia, and
acute glomerulonephritis.
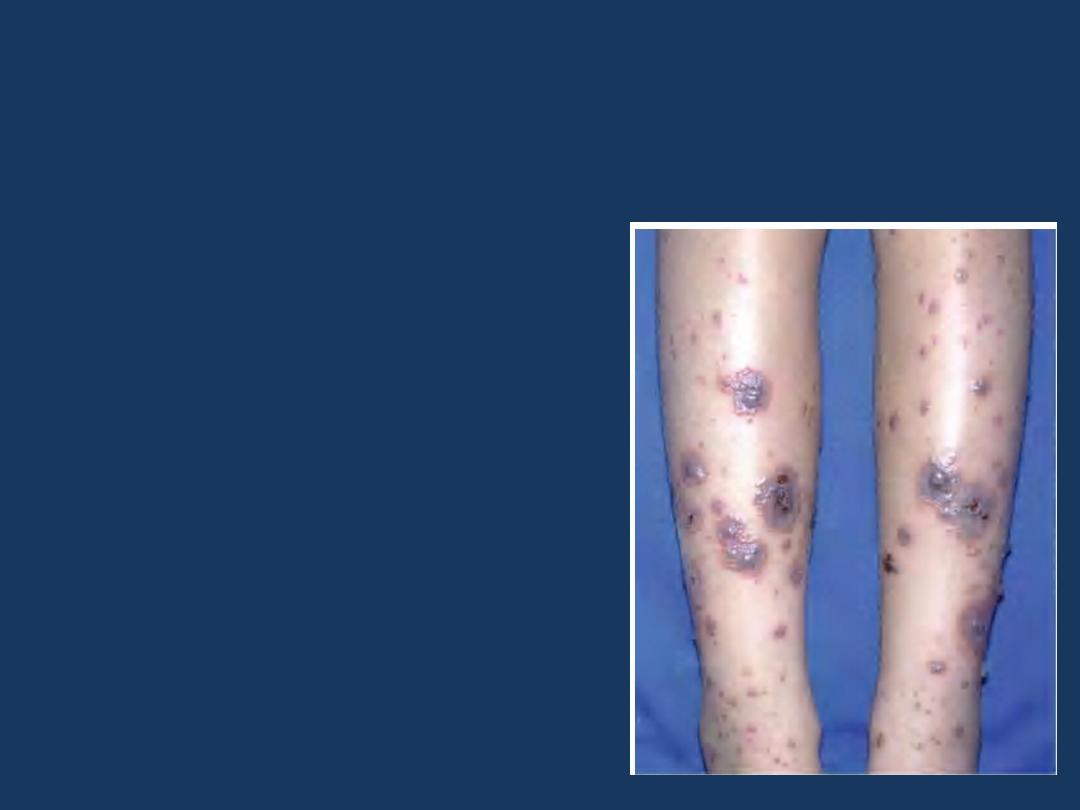
•
Henoch-Schönlein purpura
is characterized by a
purpuric rash, colicky abdominal pain, polyarthralgia,
and acute glomerulonephritis.
The involved
mechanism
is
due to the deposition of
circulating immune
complexes within vessels
throughout the body and
within the glomerular
mesangial regions.
It is an
Ig A
-mediated
vasculitis.
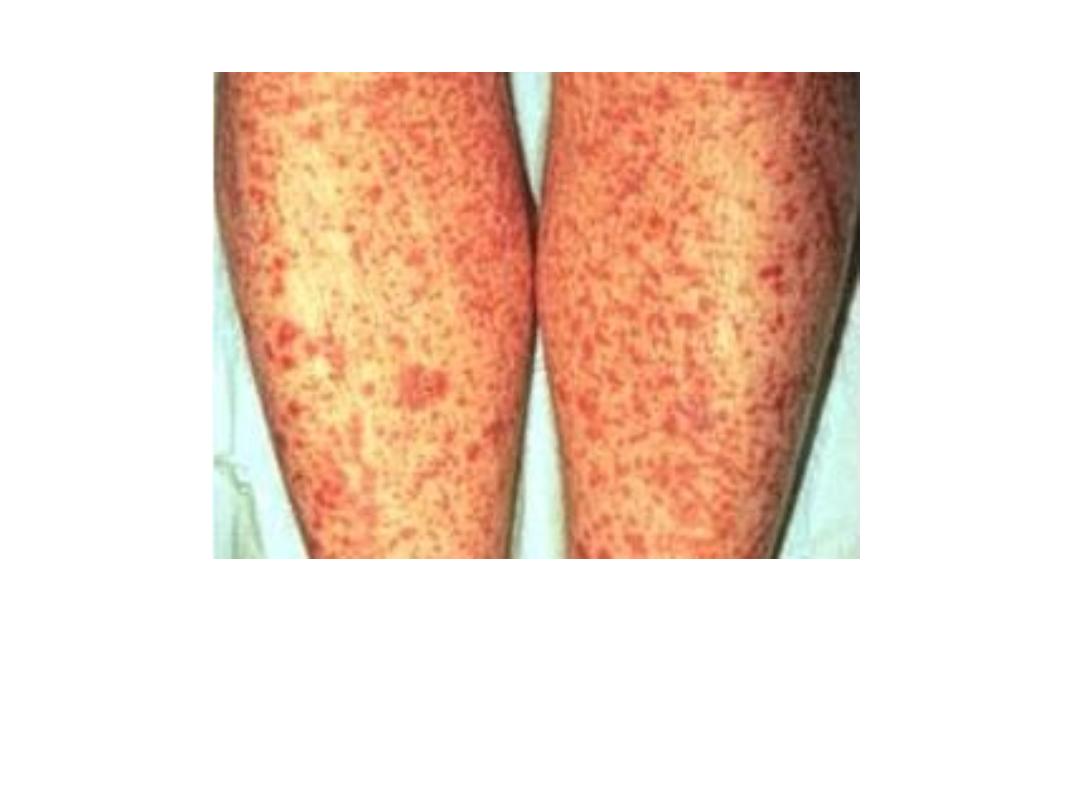
Purpura

•
All these changes result from the deposition
of circulating immune complexes within
vessels throughout the body and within the
glomerular mesangial regions.
•
•
It is an IgA mediated vasculitis.

•
•
Hereditary hemorrhagic telangiectasia
is an autosomal dominant disorder
characterized by dilated, tortuous blood
vessels with thin walls that bleed readily.
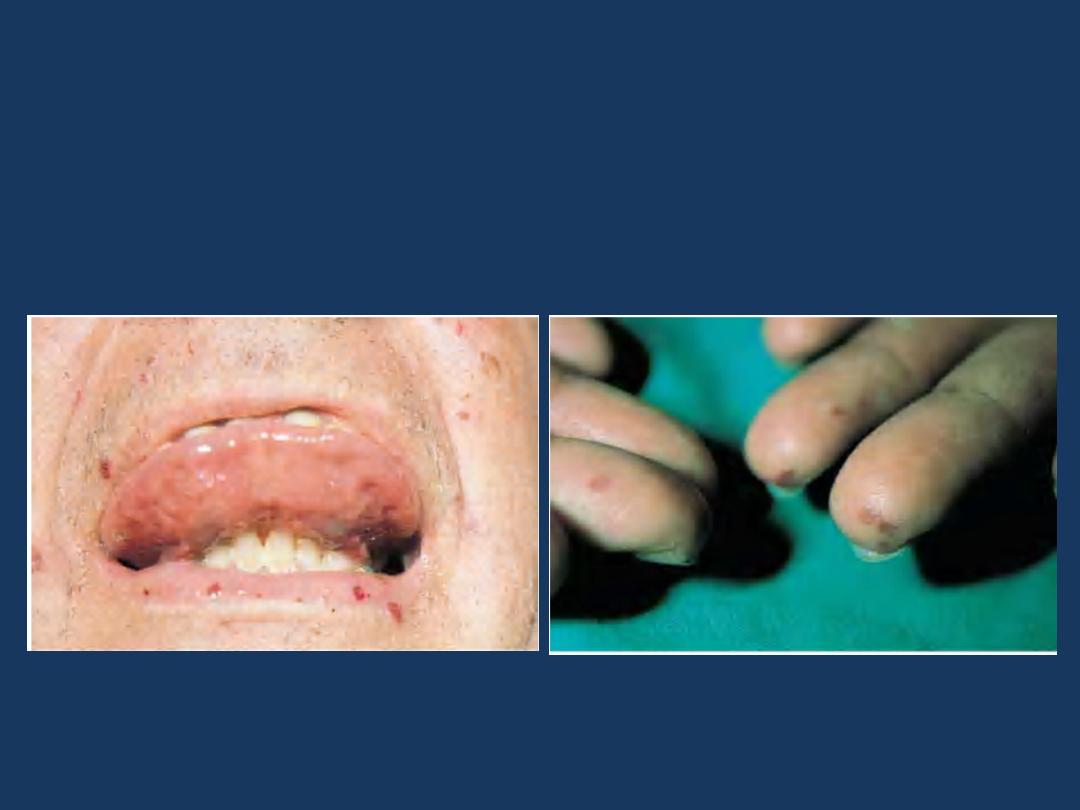
•
Hereditary hemorrhagic telangiectasia
is an
autosomal dominant disorder characterized by
dilated, tortuous blood vessels with thin walls
that bleed readily.
Hereditary hemorrhagic
telangiectasia: sublingual and labial
telangiectasia
Hereditary hemorrhagic
telangiectasia: acral
telangiectasias
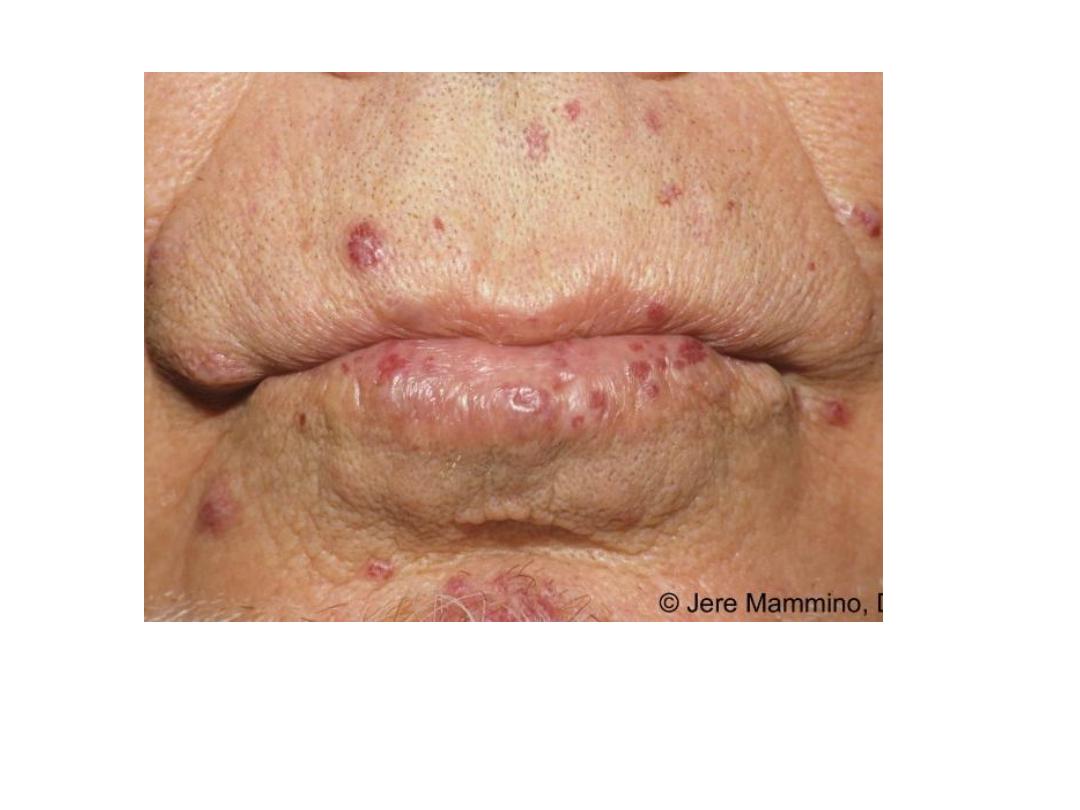
Hereditary hemorrhagic
telangiectasia


THROMBOCYTOPENIA:
Bleeding Related to Reduced Platelet Number:

Bleeding Related to Reduced Platelet Number:
•
Thrombocytopenia: Reduction in platelet number constitutes
an important cause of generalized bleeding.
•
A count below 100,000 platelets/μL is generally considered to
•
constitute thrombocytopenia.
•
However, spontaneous bleeding does not become evident
•
until platelet counts fall below 20,000 platelets/μL.
•
Platelet counts in the range of 20,000 to 50,000 platelets/μL
can aggravate post-traumatic bleeding.
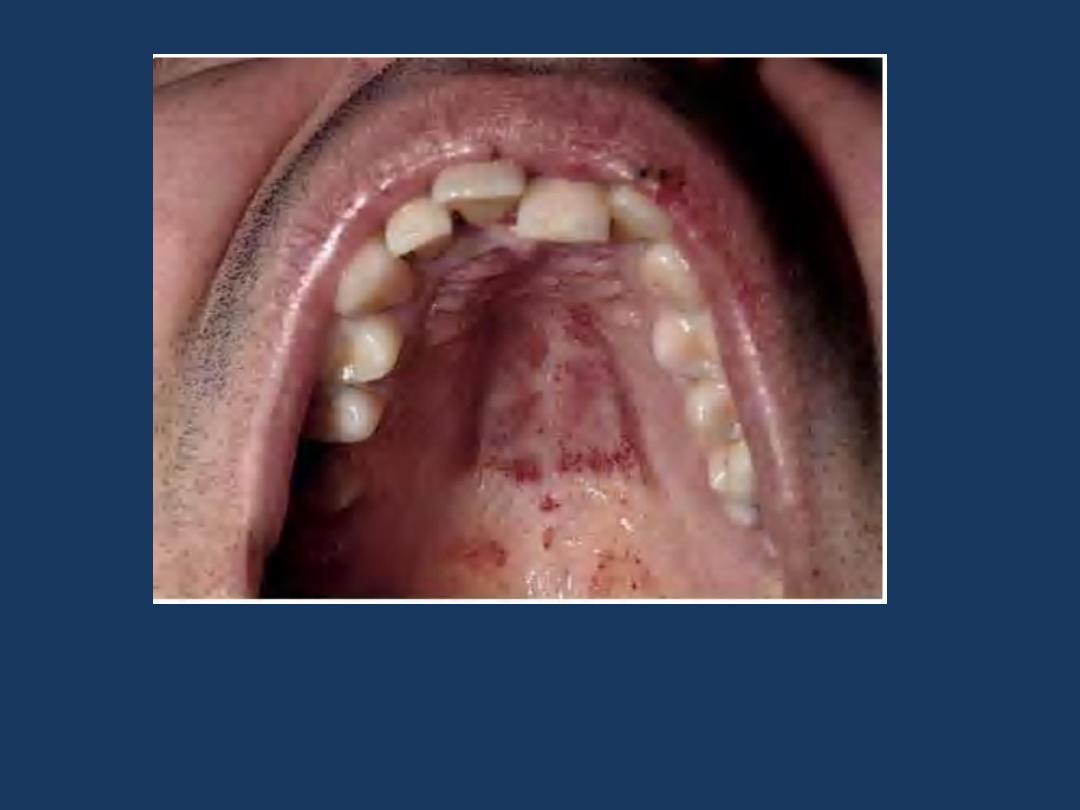
Thrombocytopenic purpura Can first manifest on the oral
mucosa or conjunctiva. Here multiple petechial hemorrhages
are seen on the palate.

•
Bleeding resulting from thrombocytopenia is
associated with a normal PT and PTT .
•
Spontaneous bleeding associated with
thrombocytopenia most often involves small vessels.
•
Common sites for such hemorrhages are the skin and
the mucous membranes of the gastrointestinal and
genitourinary tracts.
•

What are the results of screening laboratory
tests in bleeding due to thrombocytopenia?
●Platelet count is reduced.
●A prolonged bleeding time.
●A normal PT and PTT.

•
The many causes of thrombocytopenia can be
classified into the four major categories:
Decreased production of platelets:
Decreased platelet survival:
Mechanical injury:
Sequestration:
Dilutional:

Decreased production of platelets:
•
1-This can accompany generalized diseases of bone
marrow such as aplastic anemia and leukemias
•
•
2-or result from diseases that affect the
megakaryocytes somewhat selectively.
•
In vitamin B12 or folic acid deficiency, there is poor
development and accelerated destruction of
megakaryocytes within the bone marrow (ineffective
megakaryopoiesis) because DNA synthesis is impaired.

Decreased platelet survival:
•
●This important cause of thrombocytopenia
can have an
immunologic
or
nonimmunologic
etiology.

•
In the immune conditions: platelet destruction
is caused by
•
1-circulating
antiplatelet antibodies
or,
•
•
2-less often,
immune complexes.

The antiplatelet antibodies
•
1-can be directed against a self-antigen on the
platelets
(autoantibodies)
or
•
•
2-against platelet antigens that differ among
different individuals
(alloantibodies).

•
Alloimmune thrombocytopenias arise
•
when an individual is exposed to platelets of
another person, as may occur after blood
•
transfusion or during pregnancy.
•
In the latter case, neonatal or even fetal
•
thrombocytopenia occurs by a mechanism
analogous to erythroblastosis fetalis.

Nonimmunologic destruction of
platelets:
•
▪may be caused by: Mechanical injury in a
manner analogous to red cell destruction in
microangiopathic hemolytic anemia.
•
The underlying conditions are also similar,
including prosthetic heart valves and diffuse
narrowing of the microvessels (e.g., malignant
hypertension).

•
●
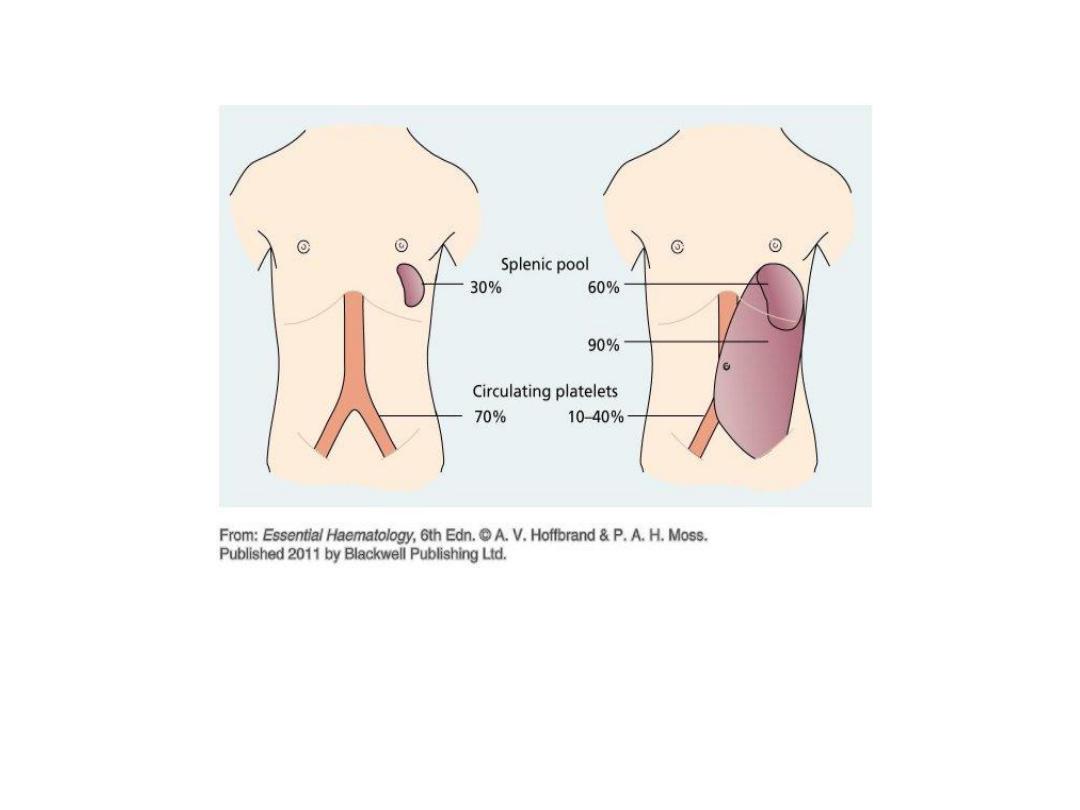
Sequestration:
Thrombocytopenia, usually
moderate in severity, may develop in any
patient with marked splenomegaly, a
condition sometimes referred to as
hypersplenism.

•
The spleen normally sequesters 30% to 40% of
the body's platelets, which remain in
equilibrium with the circulating pool.
•
When necessary, hypersplenic
thrombocytopenia can be ameliorated by
splenectomy.

•
●Dilutional: Massive transfusions can produce
a dilutional thrombocytopenia.
•
Blood stored for longer than 24 hours
contains virtually no viable platelets; thus,
plasma volume and red cell mass are
reconstituted by transfusion, but the number
of circulating platelets is relatively reduced.

Immune Thrombocytopenic Purpura
(ITP):
•
ITP can occur in:
•
•The setting of a variety of conditions and
exposures
(secondary ITP)
or
•
•In the absence of any known risk factors
(primary or idiopathic ITP).

primary ITP
•
There are two clinical subtypes of primary ITP:
•
1-
acute
and
•
2-
chronic
;
•
both are autoimmune disorders in which
platelet destruction results from the
formation of antiplatelet autoantibodies.

Chronic ITP:
•
Pathogenesis: Chronic ITP is caused by the
formation of autoantibodies against platelet
membrane glycoproteins.

•
Antibodies reactive with these membrane
glycoproteins can be demonstrated in the
plasma as well as bound to the platelet
surface (platelet associated immunoglobulins)
in approximately 80% of patients.
•
In the overwhelming majority of cases, the
antiplatelet antibodies are of the
IgG
class.
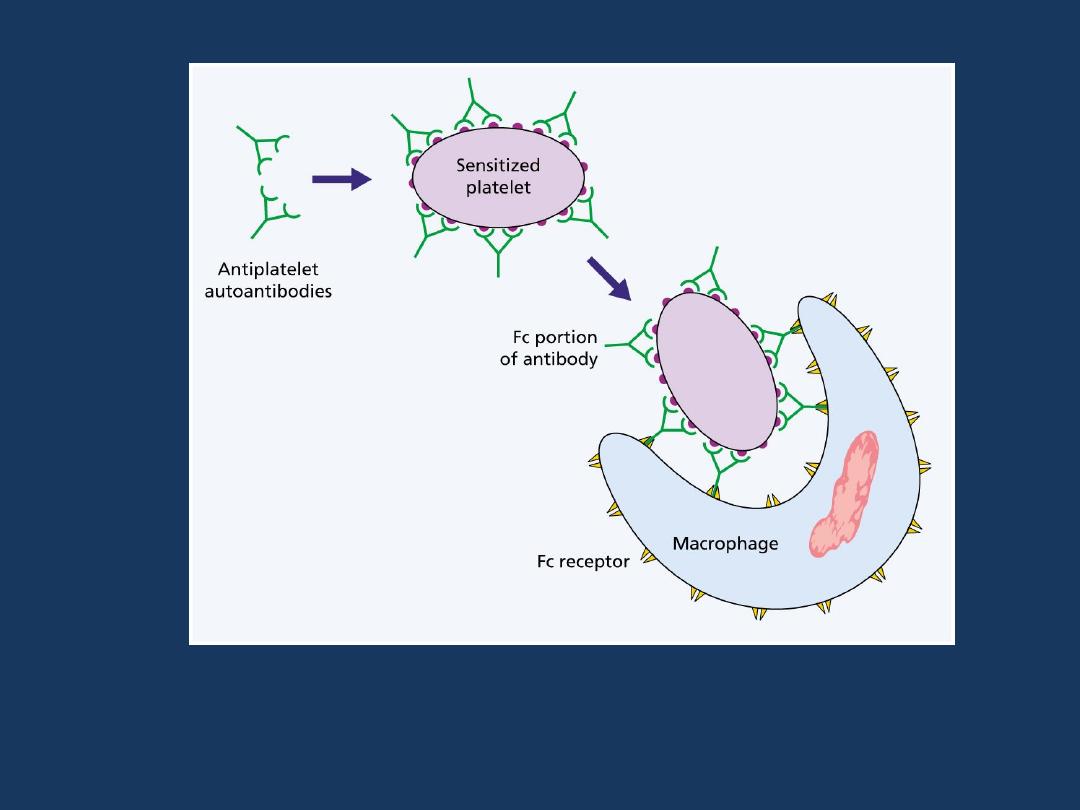
Chronic ITP:
Pathogenesis
Chronic ITP is caused by the formation of
autoantibodies against
platelet membrane glycoproteins. In the majority of cases, the
antiplatelet antibodies are
of the IgG class.

Chronic ITP:
•
The mechanism of platelet destruction is as
follows: Opsonized platelets are rendered
susceptible to phagocytosis by the cells of the
mononuclear phagocyte system especially of
the spleen.

•
About 75% to 80% of patients are remarkably
improved after splenectomy, indicating that
the spleen is the major site of removal of
sensitized platelets.
•
•
Since it is also an important site of
•
autoantibody synthesis, the beneficial effects
of splenectomy may in part derive from
•
removal of the source of autoantibodies.

Acute ITP
•
Like chronic ITP, this condition is caused by
antiplatelet autoantibodies, but its clinical
features and course are distinct.
•
Acute ITP is a disease of childhood occurring
•
with equal frequency in both sexes.

Q.3-Which one of the following laboratory
determinations is
abnormally prolonged
in
idiopathic thrombocytopenic purpura?
a. Partial thromboplastin time (PTT).
b. Bleeding time.
c. Coagulation time.
d. Prothrombin time (PT).
e. Thrombin time.

Q.3-Which one of the following laboratory
determinations is
abnormally prolonged
in
idiopathic thrombocytopenic purpura?
a. Partial thromboplastin time (PTT).
b. Bleeding time.
c. Coagulation time.
d. Prothrombin time (PT).
e. Thrombin time.

Drug-induced immune
thrombocytopenia:
•
An immunological mechanism has been
•
demonstrated as the cause of many drug-
induced thrombocytopenias.
•
Quinine, quinidine and heparin are
particularly common causes.

•
An antibody-drug-protein complex is deposited
on the platelet surface.
•
If complement is attached and the sequence goes
to completion, the platelet may be lysed directly.
•
Otherwise, it is removed by reticuloendothelial
cells because of opsonization with
immunoglobulin and / or the C3
•
component of complement.
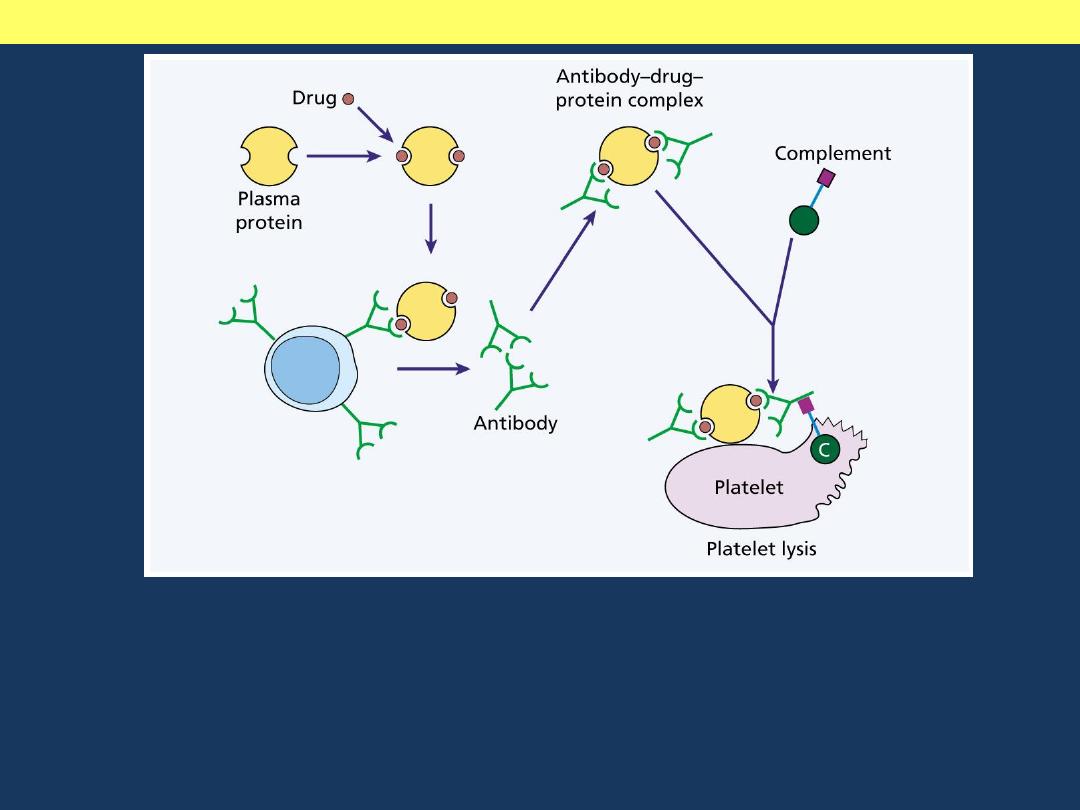
Mechanisms of drug induced thrombocytopenia
An antibody-drug-protein complex is deposited on the platelet surface. If
complement is attached and the sequence goes to completion, the platelet
may be lysed directly. Otherwise, it is removed by reticuloendothelial
cells because of opsonization with immunoglobulin and / or the C3
component of complement.

•
The platelet count is often less than 10 x
109/L, and the bone marrow shows normal or
increased numbers of megakaryocytes.
•
Drug dependent antibodies against platelets
may be demonstrated in the sera of some
patients.

Q.4-For the past 6 months, a 35-year-old female has had
excessively heavy menstrual flow. She has also noticed
increasing numbers of pinpoint hemorrhages on her lower
extremities in the past month. Physical examination reveals
no organomegaly or lymphadenopathy. A complete blood
count (CBC) shows a
hemoglobin concentration of 14.2 g/
dL
,
platelet count of 19,000/µL
, and
white blood cell count of
6000/µL
.
The most likely basis of her bleeding tendency is:
a. Abnormalities in production of platelets by
megakaryocytes.
b. Destruction of antibody-coated platelets by the spleen.
c. Excessive loss of platelets in menstrual blood.
d. Suppression of pluripotent stem cells.
e. Defective platelet-endothelial interactions.

Q.4-For the past 6 months, a 35-year-old female has had
excessively heavy menstrual flow. She has also noticed
increasing numbers of pinpoint hemorrhages on her lower
extremities in the past month. Physical examination
reveals no organomegaly or lymphadenopathy. A complete
blood count (CBC) shows a
hemoglobin concentration of
14.2 g/ dL
,
platelet count of 19,000/µL
, and
white blood
cell count of 6000/µL
.
The most likely basis of her bleeding
tendency is:
a. Abnormalities in production of platelets by
megakaryocytes.
b. Destruction of antibody-coated platelets by the spleen
.
c. Excessive loss of platelets in menstrual blood.
d. Suppression of pluripotent stem cells.
e. Defective platelet-endothelial interactions.

Bleeding Disorders Related To
Defective Platelet Functions:
•
Qualitative defects of
•
platelet function can be
•
1-congenital or
•
2-acquired.

•
Several congenital disorders characterized
•
by prolonged bleeding time and normal platelet count
have been described.
•
Congenital disorders of platelet function can be
classified into three groups on the basis of the specific
functional abnormality:
•
1. Defects of adhesion.
•
2. Defects of aggregation.
•
3. Disorders of platelet secretion (release reaction).

Acquired defects of platelet function:
•
▪Ingestion of aspirin and other nonsteroidal anti-
inflammatory drugs which significantly
prolongs the bleeding time.
•
▪Aspirin: Is a potent, irreversible inhibitor of the
enzyme cyclooxygenase.
•
▪Uremia: Several abnormalities of platelet
function are found.

BREAK

Hemorrhagic Diatheses Related To
Abnormalities In Clotting Factors:
•
A deficiency of every clotting factor has been
reported to be the cause of a bleeding
disorder, with the exception of factor XII
deficiency, which does not cause bleeding.

•
The bleeding in factor deficiencies differs from platelet
deficiencies in that spontaneous petechiae or purpura
are uncommon.
•
Rather, the bleeding is manifested by large post
traumatic ecchymoses or hematomas, or prolonged
bleeding after a laceration or any form of surgical
procedure.
•
Bleeding into the gastrointestinal and urinary tracts,
and particularly into weight-bearing joints, is common.
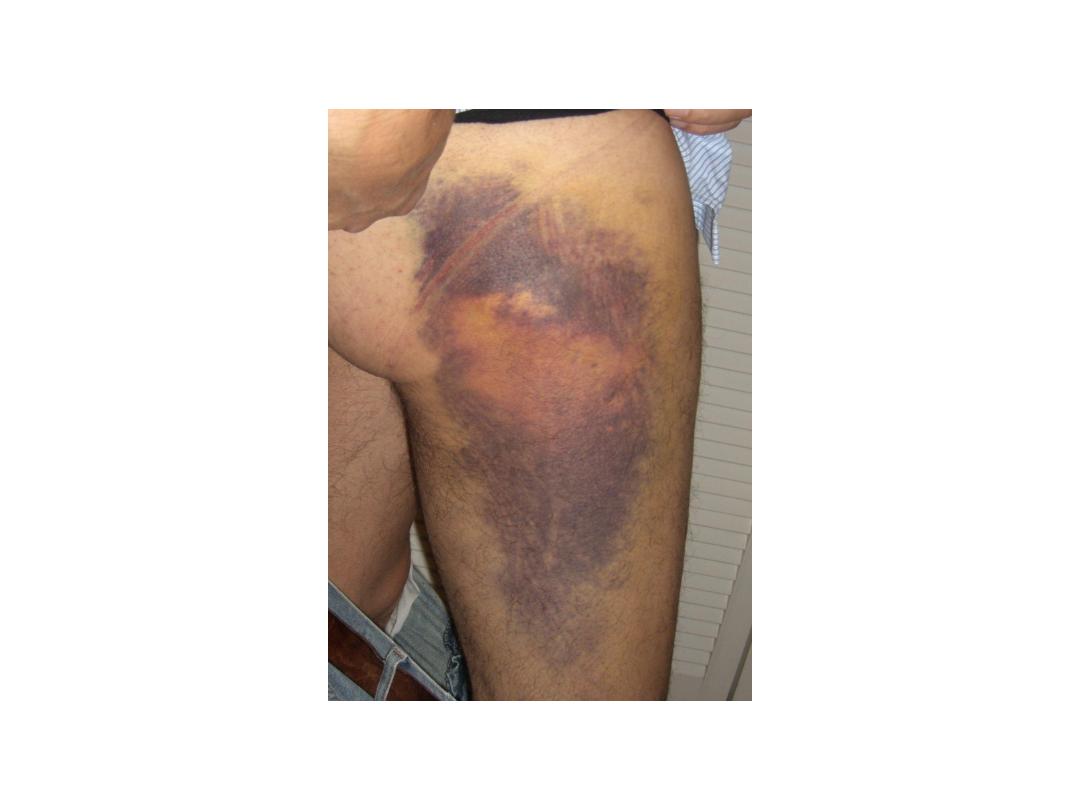

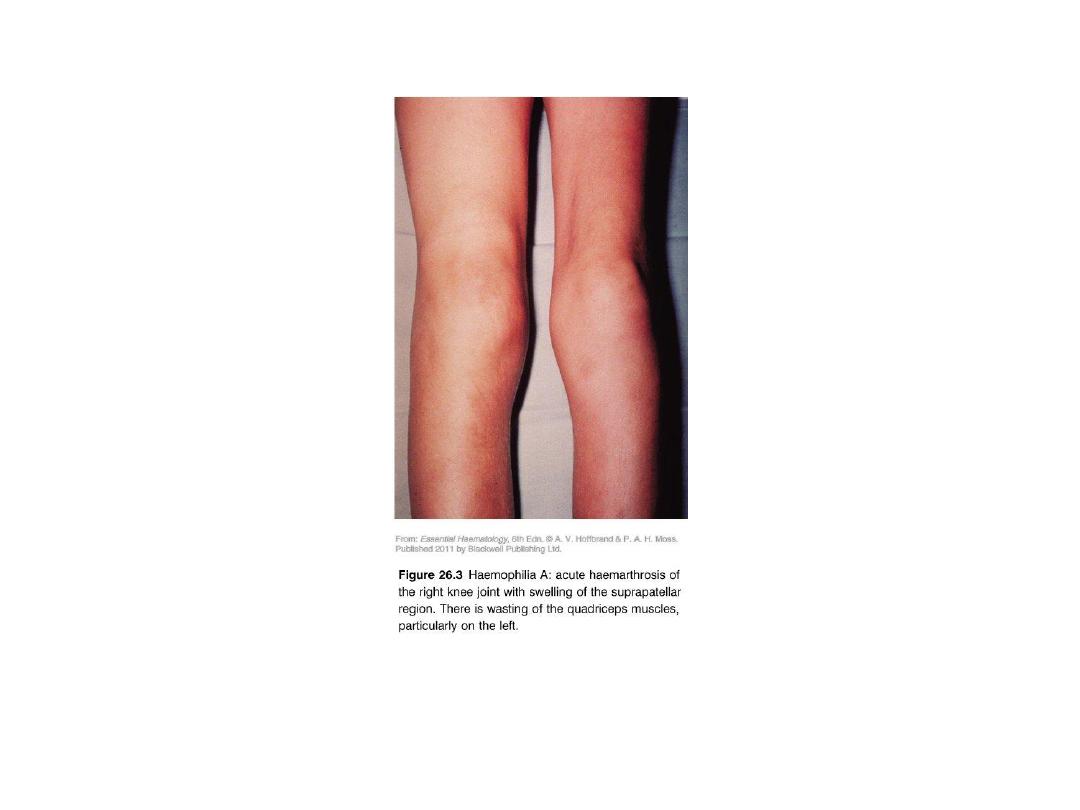

•
Hereditary deficiencies have been identified for each of the
clotting factors.
•
Deficiencies of factor VIII (hemophilia A) and of factor IX
(Christmas disease, or hemophilia B) are
•
transmitted as sex-linked recessive disorders.
•
Most others follow autosomal patterns of
•
transmission.
•
These hereditary disorders typically involve a single clotting
factor.

Deficiencies of Factor VIII-vWF
Complex:
•
Hemophilia A and von Willebrand disease,
•
two of the most common inherited disorders
of bleeding, are caused by qualitative or
•
quantitative defects involving the factor VIII-
vWF complex.
•
Plasma factor VIII-vWF is a complex made up
of two separate proteins (factor VIII and
vWF)..

Deficiencies of Factor VIII-vWF
Complex:
cont.
•
Factor VIII; is an intrinsic pathway component
required for activation of factor X.
•
Deficiency of factor VIII gives rise to
hemophilia A
•
Circulating factor VIII is noncovalently
associated with very large vWF multimers.

Deficiencies of Factor VIII-vWF
Complex:
cont.
•
The most important function of vWF in vivo is
to promote the adhesion of platelets to
subendothelial matrix.
•
The two components of the factor VIII-vWF
•
complex are encoded by separate genes and
synthesized in different cells.
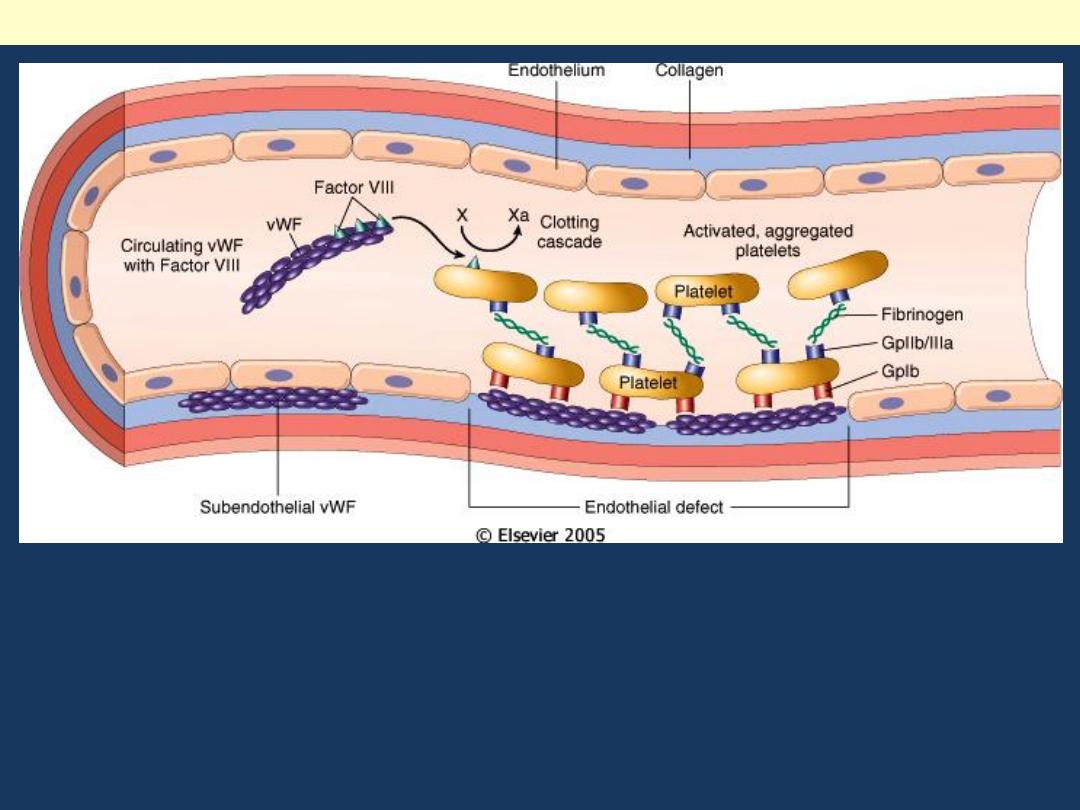
Circulating factor VIII is noncovalently associated with
very large vWF multimers. The most important function
of vWF in vivo is to promote the adhesion of platelets to
subendothelial matrix.
Structure and function of factor VIII-von Willebrand factor (vWF) complex
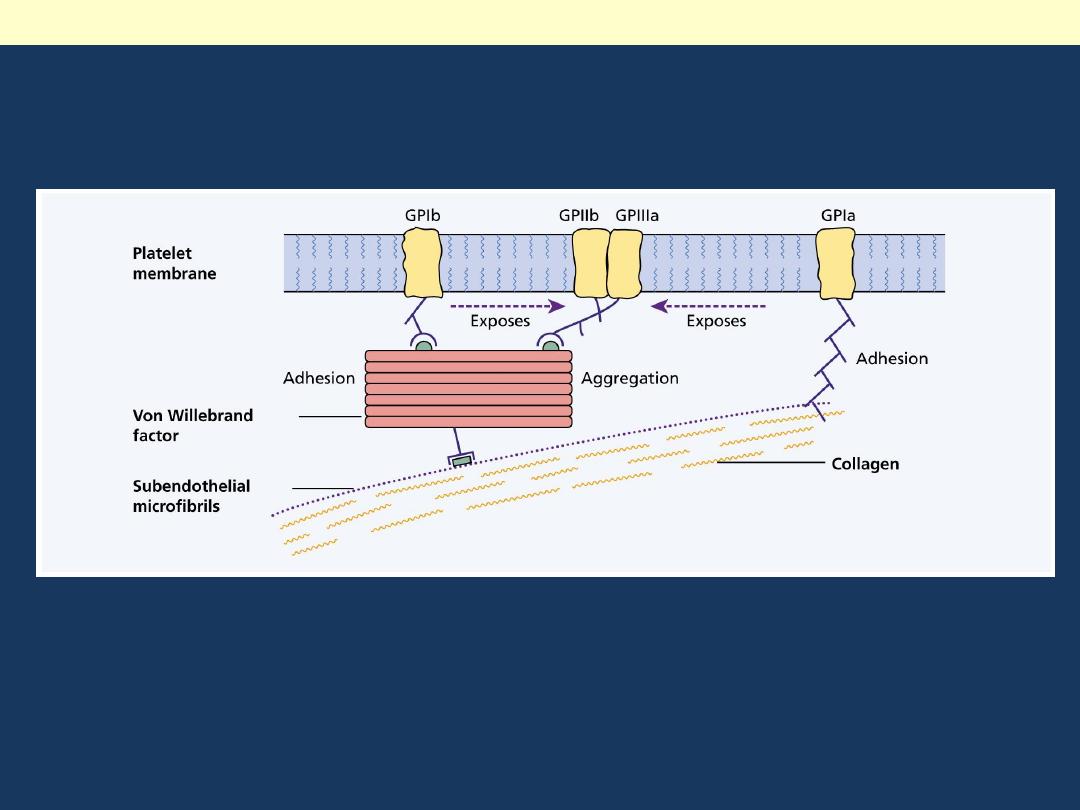
Structure and function of factor VIII-von Willebrand factor (vWF) complex

Deficiencies of Factor VIII-vWF
Complex:
cont.
•
vWF is produced by endothelial cells and
megakaryocytes and can be demonstrated in
platelet α- granules.
•
Endothelial cells are the major source of
subendothelial and plasma vWF.
•
vWF gene is located on chromosome 12.

Deficiencies of Factor VIII-vWF
Complex:
cont.
•
Factor VIII is made in several tissues;
sinusoidal endothelial cells and Kupffer cells in
the liver and glomerular and tubular epithelial
cells in the kidney appear to be particularly
important sites of synthesis.
•
Factor VIII gene is located on X chromosome

Von Willebrand Disease:
•
With an estimated frequency of 1%, von
Willebrand disease is believed to be one of
the most common inherited disorders of
bleeding in humans.
•
Clinically, it is characterized by spontaneous
bleeding from mucous membranes, excessive
bleeding from wounds, menorrhagia.

Von Willebrand Disease:
cont.
•
In this disorder there is either a reduced level
or abnormal function of VWF resulting from a
point mutation or major deletion.
•
Patients with von Willebrand disease have
defects in platelet function despite a normal
•
platelet count.

Lab findings VW disease:
•
Lab findings:
•
Patients with von Willebrand disease typically have:
•
•A prolonged bleeding time.
•
•A normal platelet count.
•
•The plasma level of active vWF is reduced.
•
(Because vWF stabilizes factor VIII by binding to it, a
deficiency of vWF gives rise to a
•
secondary decrease in factor VIII levels); this may be
reflected by a prolongation of the
•
PTT in von Willebrand disease types 1 and 3.

VW disease
cont.
•
In most cases, it is transmitted as an
autosomal dominant disorder, but several rare
autosomal recessive variants have been
identified.
•
Because a severe deficiency of vWF has a
marked affect on the stability of factor VIII,
some of the bleeding characteristics resemble
those seen in hemophilia.

Q.5-A young adult patient has just been diagnosed with
Von Willebrand disease.
Which of the following statements should you make to
advise the patient of potential consequences of this
disease?
a. You may need an allogeneic bone marrow transplant.
b. Expect increasing difficulties with joint mobility.
c. Anticoagulation is needed to prevent deep venous
thrombosis.
d. You may have excessive bleeding following tooth
extraction.
e. A splenectomy may be necessary to control the
disease.

Q.5-A young adult patient has just been diagnosed with
Von Willebrand disease.
Which of the following statements should you make to
advise the patient of potential consequences of this
disease?
a. You may need an allogeneic bone marrow transplant.
b. Expect increasing difficulties with joint mobility.
c. Anticoagulation is needed to prevent deep venous
thrombosis.
d. You may have excessive bleeding following tooth
extraction.
e. A splenectomy may be necessary to control the
disease.

Hemophilia A (Factor VIII Deficiency):
•
Hemophilia A is the most common hereditary
disease associated with serious bleeding.
•
It is caused by a reduction in the amount or
activity of factor VIII.
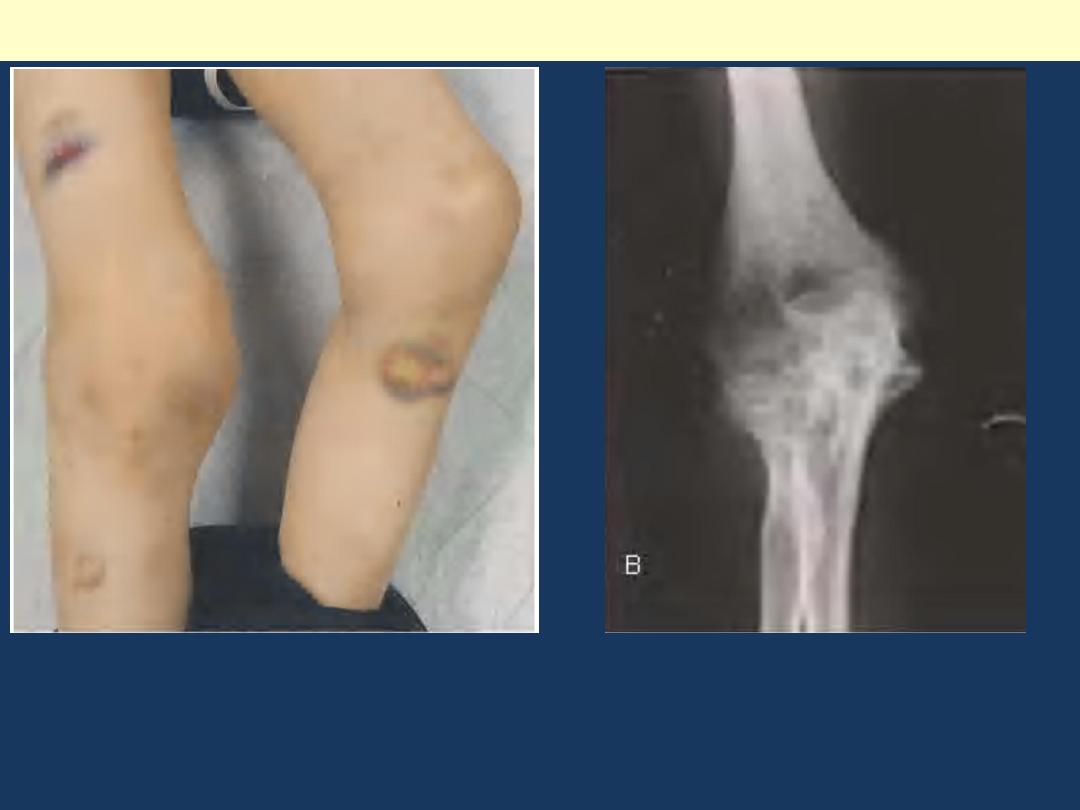
Hemophilia A: Hemarthrosis
Chronic right knee hemarthrosis with fresh and fading ecchymoses on legs.
Radiological image of knee showing loss of joint space with apparent fusion of
femoral and tibial articulation and cystic changes.

Hemophilia A (Factor VIII Deficiency):
•
Hemophilia A is inherited as an X-linked recessive trait,
and thus occurs in males and in homozygous females.
•
However, excessive bleeding has been described in
heterozygous females, presumably due to extremely
unfavorable lyonization (inactivation of the normal X
chromosome in most of the cells).
•
Approximately 30% of patients have no family history;
their disease is presumably caused by new mutations.

Hemophilia A (Factor VIII Deficiency):
•
Hemophilia A exhibits a wide range of clinical
severity that correlates well with the level of
factor VIII activity.
•
•Those with less than 1% of normal activity
develop severe disease.
•
•Levels between 2% and 5% of normal are
associated with moderate disease.
•
•Patients with 6% to 50% of activity develop mild
disease.

Hemophilia A (Factor VIII Deficiency):
•
The variable degrees of factor VIII deficiency
are largely explained by heterogeneity in
•
the causative mutations.
•
Several genetic lesions (deletions, nonsense
mutations that create stop codons, splicing
errors) have been documented.

Hemophilia A (Factor VIII Deficiency):
•
Lab findings:
•
Patients with hemophilia A typically have:
•
•A normal bleeding time.
•
•A normal platelet count, and a normal PT.
•
•A prolonged PTT.
•
(These tests point to an abnormality of the
intrinsic coagulation pathway).
•
►Factor VIII-specific assays are required for
diagnosis.

Q.6-A 13-year-old male has less than 1%
factor VIII activity measured in plasma. If
he does not receive transfusions of factor
VIII concentrate,
which of the following
manifestations of this deficiency is most
likely to ensue?
a. Splenomegaly.
b. Conjunctival petechiae.
c. Hemolysis.
d. Hemochromatosis.
e. Hemarthroses.

Q.6-A 13-year-old male has less than 1%
factor VIII activity measured in plasma. If
he does not receive transfusions of factor
VIII concentrate,
which of the following
manifestations of this deficiency is most
likely to ensue?
a. Splenomegaly.
b. Conjunctival petechiae.
c. Hemolysis.
d. Hemochromatosis.
e. Hemarthroses.

Hemophilia B (Christmas Disease, Factor IX
Deficiency):
•
Severe factor IX deficiency produces a
disorder clinically indistinguishable from
factor VIII deficiency (hemophilia A).
•
This should not be surprising, given that factor
VIII and IX function together to activate factor
X.

Hemophilia B (Christmas Disease,
Factor IX Deficiency):
•
Wide spectrums of mutations involving the
factor IX gene are found in hemophilia B.
•
Like hemophilia A, it is inherited as an Xlinked
recessive trait and shows variable clinical
severity.
•
In about 14% of these patients, factor IX is
present but nonfunctional.

Hemophilia B (Christmas Disease,
Factor IX Deficiency):
•
Lab findings:
•
Patients with hemophilia B typically have:
•
•A normal bleeding time.
•
•A normal platelet count, and a normal PT.
•
•A prolonged PTT.
•
►Factor IX-specific assays are required for
diagnosis.

Q.7-All of the following conditions are
associated with a prolonged bleeding time
EXCEPT:
a. von Willebrand disease.
b. Deficiency of factor IX.
c. Long-term treatment with aspirin.
d. Idiopathic thrombocytopenic purpura.
e. Defect in platelet adhesion.

Q.7-All of the following conditions are
associated with a prolonged bleeding time
EXCEPT:
a. von Willebrand disease.
b. Deficiency of factor IX.
c. Long-term treatment with aspirin.
d. Idiopathic thrombocytopenic purpura.
e. Defect in platelet adhesion.

Disseminated Intravascular
Coagulation (DIC):
•
DIC is an acute, subacute, or chronic
thrombohemorrhagic disorder occurring as a
•
secondary complication in a variety of
diseases.

Disseminated Intravascular
Coagulation (DIC):
•
It is characterized by activation of the
coagulation sequence that leads to the
formation of microthrombi throughout the
microcirculation of the body, often in a
quixotically uneven distribution.
•
●Sometimes the coagulopathy is localized to a
specific organ or tissue.

Disseminated Intravascular
Coagulation (DIC):
•
As a consequence of the thrombotic diathesis,
there is consumption of platelets, fibrin,
•
and coagulation factors and, secondarily,
activation of fibrinolytic mechanisms.

Disseminated Intravascular
Coagulation (DIC):
•
Thus, DIC can present with signs and symptoms relating to:
•
▪Tissue hypoxia and infarction caused by the myriad
microthrombi or
•
▪A hemorrhagic disorder related to depletion of the
elements required for hemostasis (hence, the term
"consumption coagulopathy" is sometimes used to
•
describe DIC).
•
Activation of the fibrinolytic mechanism aggravates the
hemorrhagic diathesis.

Disseminated Intravascular
Coagulation (DIC):
•
Etiology and Pathogenesis: At the outset, it
must be emphasized that DIC is not a primary
•
disease. It is a coagulopathy that occurs in the
course of a variety of clinical conditions.

DIC triggering mechanisms
•
Two major mechanisms trigger DIC:
•
1. Release of tissue factor or thromboplastic
substances into the circulation:
•
2. Widespread injury to the endothelial cells:

Disseminated Intravascular
Coagulation (DIC):
•
1. Release of tissue factor or thromboplastic substances into the circulation:
•
Tissue thromboplastic substances can be derived from a variety of sources, such as
a-the placenta in obstetric complications and
•
b- the granules of leukemic cells in acute promyelocytic leukemia.
•
c- Mucus released from certain adenocarcinomas can also act as a thromboplastic
•
substance by directly activating factor X, independent of factor VII.
•
d- In gram-negative sepsis (an important cause of DIC), bacterial endotoxins cause
activated monocytes to release interleukin-1 and TNF, both of which increase the
expression of tissue factor on endothelial cell membranes and simultaneously
decrease the expression of
•
thrombomodulin.
The net result is a shift in balance toward procoagulation.

Disseminated Intravascular
Coagulation (DIC):
•
2. Widespread injury to the endothelial cells:
The other major trigger,
•
can initiate DIC by causing release of tissue
factor, promoting platelet aggregation, and
activating the intrinsic coagulation pathway.
•
TNF is an extremely important mediator of
endothelial cell inflammation and injury in
septic shock.

Disseminated Intravascular
Coagulation (DIC):
•
Even subtle endothelial injury can unleash
procoagulant activity by enhancing membrane
expression of tissue factor.
•
Widespread endothelial injury may be produced
by deposition of antigen-antibody complexes
(e.g., systemic lupus erythematosus),
temperature extremes (e.g., heat stroke, burns),
or microorganisms (e.g., meningococci,
rickettsiae).
•
The initiating factors in these conditions are often
multiple and interrelated.
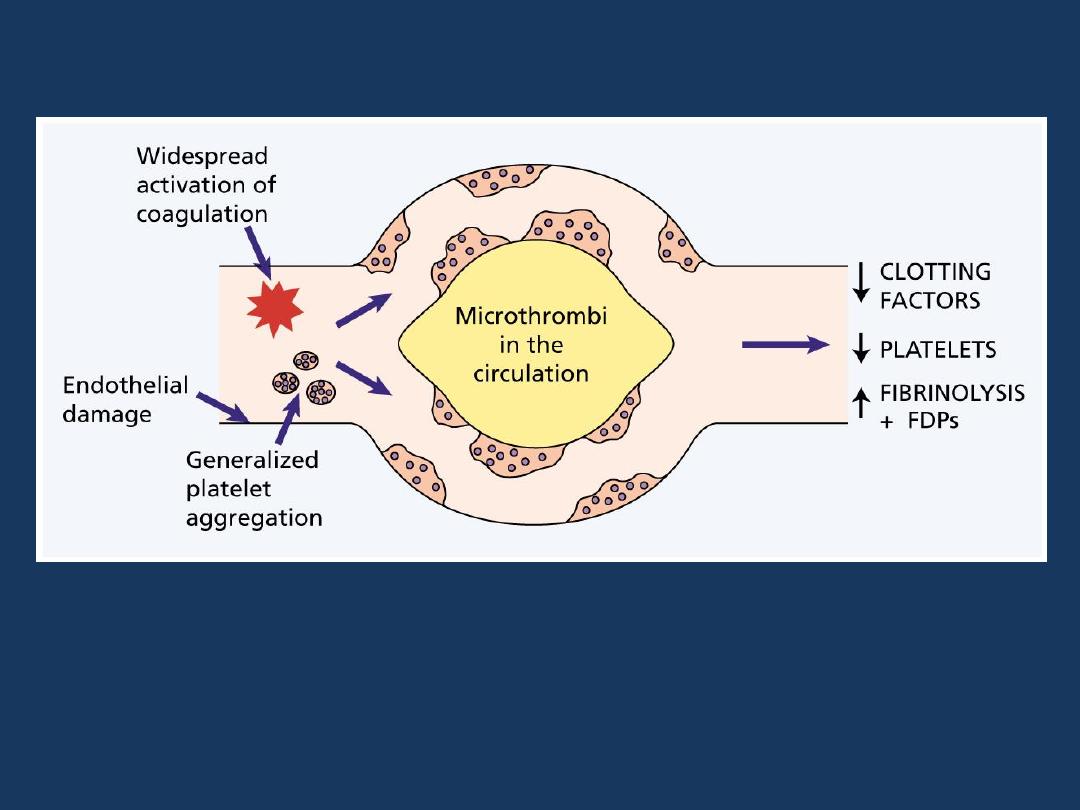
Disseminated Intravascular Coagulation (DIC)
The pathogenesis of disseminated intravascular
coagulation and the changes in clotting factors, platelets
and fibrin degradation products (FDPs) that occur in this
syndrome.

Disseminated Intravascular
Coagulation (DIC):
•
The consequences of DIC are twofold:
•
1-Thrombotic diathesis
•
2- Haemmorhagic diathesis

Disseminated Intravascular
Coagulation (DIC):
•
1-Thrombotic diathesis
•
There is widespread deposition of fibrin within
the microcirculation. This can lead to:
•
▪Ischemia of the more severely affected or
more vulnerable organs
•
▪A hemolytic anemia resulting from
fragmentation of red cells as they squeeze
through the narrowed microvasculature
(microangiopathic hemolytic anemia).

Disseminated Intravascular
Coagulation (DIC):
•
2- Haemmorhagic diathesis
•
A hemorrhagic diathesis can dominate the clinical
picture.
This results from consumption of platelets and clotting
factors as well as activation of plasminogen.
•
Plasmin can not only cleave fibrin, but also digest
factors V and VIII, thereby reducing their concentration
further.

Disseminated Intravascular
Coagulation (DIC):
•
Morphology: In general, thrombi are found in
the following sites in decreasing order of
•
frequency: brain, heart, lungs, kidneys,
adrenals, spleen, and liver.
•
However, no tissue is spared, and thrombi are
occasionally found in only one or several
•
organs without affecting others.
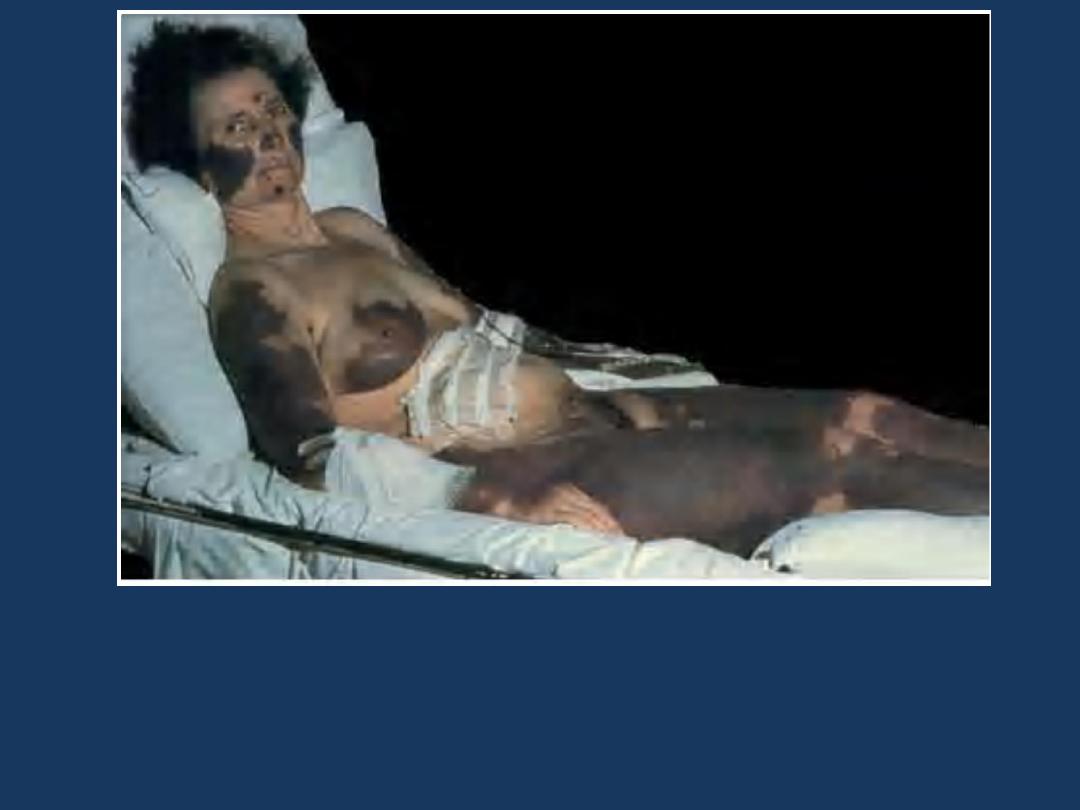
Disseminated intravascular coagulation: purpura fulminans:
Extensive geographic areas of cutaneous infarction with
hemorrhage involving the face, breast, and extremities.

Q.8-Well-known causes of disseminated
intravascular coagulation (DIC) include all
of the following conditions
EXCEPT:
a. Retained dead fetus.
b. Prostatic carcinoma.
c. Hemolytic transfusion reaction.
d. Gram-negative sepsis.
e. Heparin administration.

Q.8-Well-known causes of disseminated
intravascular coagulation (DIC) include all
of the following conditions
EXCEPT:
a. Retained dead fetus.
b. Prostatic carcinoma.
c. Hemolytic transfusion reaction.
d. Gram-negative sepsis.
e. Heparin administration.

Acquired disorders
•
Acquired disorders are usually characterized by
multiple clotting abnormalities.
•
1-Vitamin K deficiency: Results in impaired synthesis of
factors II, VII, IX, and X and protein C.
•
2-Since the liver makes virtually all the clotting factors:
•
Severe parenchymal liver disease: Can be associated
•
with a hemorrhagic diathesis.
•
3-Disseminated intravascular coagulation: Produces a
deficiency of multiple coagulation factors.

Q.9-A 45-year-old female has chronic hepatitis C
infection with serum concentrations of (GOT) of 310
U/L, (GPT) of 275 U/L, total bilirubin of 7.6 mg/dL,
direct bilirubin of 5.8 mg/dL, ALP of 75 U/L.
Which of the following laboratory test results for
hemostatic function is most likely to be abnormal?
a. Immunoassay for plasma von Willebrand factor.
b. Platelet count.
c. Prothrombin time (PT).
d. Fibrin split products.
e. Bleeding time.

Q.9-A 45-year-old female has chronic hepatitis C
infection with serum concentrations of (GOT) of 310
U/L, (GPT) of 275 U/L, total bilirubin of 7.6 mg/dL,
direct bilirubin of 5.8 mg/dL, ALP of 75 U/L.
Which of the following laboratory test results for
hemostatic function is most likely to be abnormal?
a. Immunoassay for plasma von Willebrand factor.
b. Platelet count.
c. Prothrombin time (PT).
d. Fibrin split products.
e. Bleeding time.

END