Bleeding disorders
F:\lectures\4th
grade\Pathology\New folder

Bleeding types
F:\lectures\4th
grade\Pathology\New folder
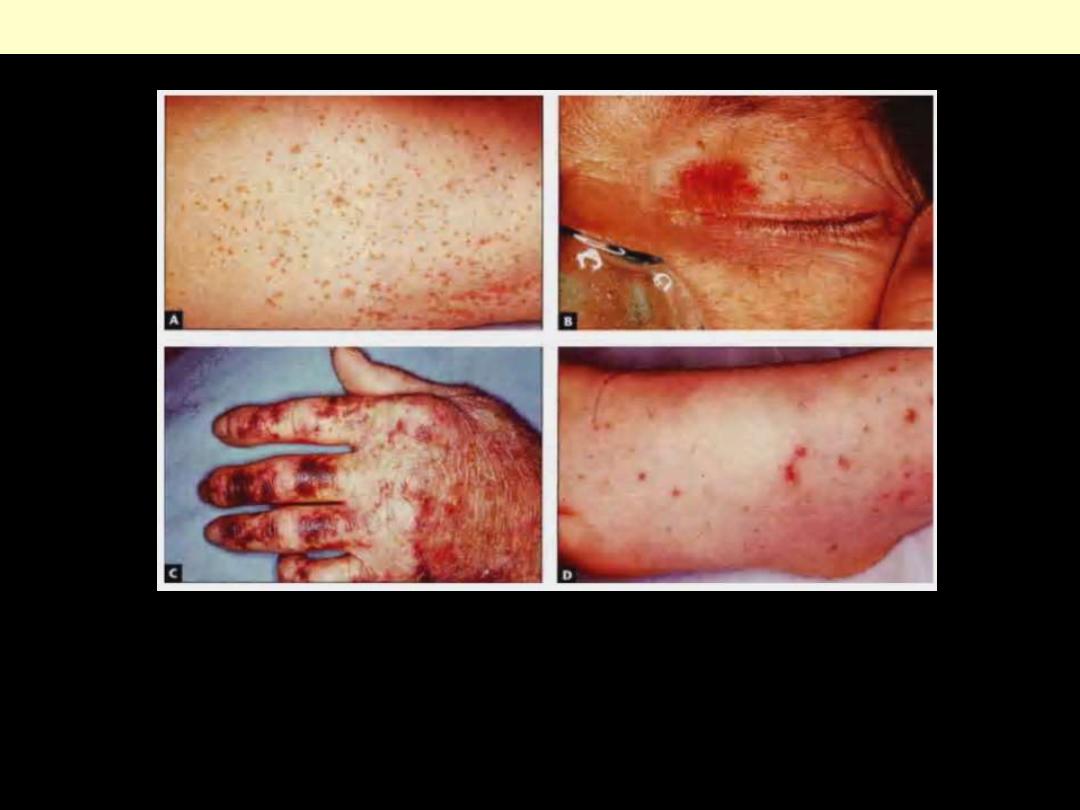
Petechiae are minute (1- to 2-mm) hemorrhages into skin, mucous membranes, or serosal surfaces.
Purpura is raised slightly larger than petechiae (3- to 5-mm) hemorrhages and can be associated with
many of the same disorders that cause petechiae.
Ecchymoses are larger (1- to 2-cm) subcutaneous hematomas (bruises)
Petechiae, Purpura & ecchymoses

Bleeding – Vessel wall
abnormalities
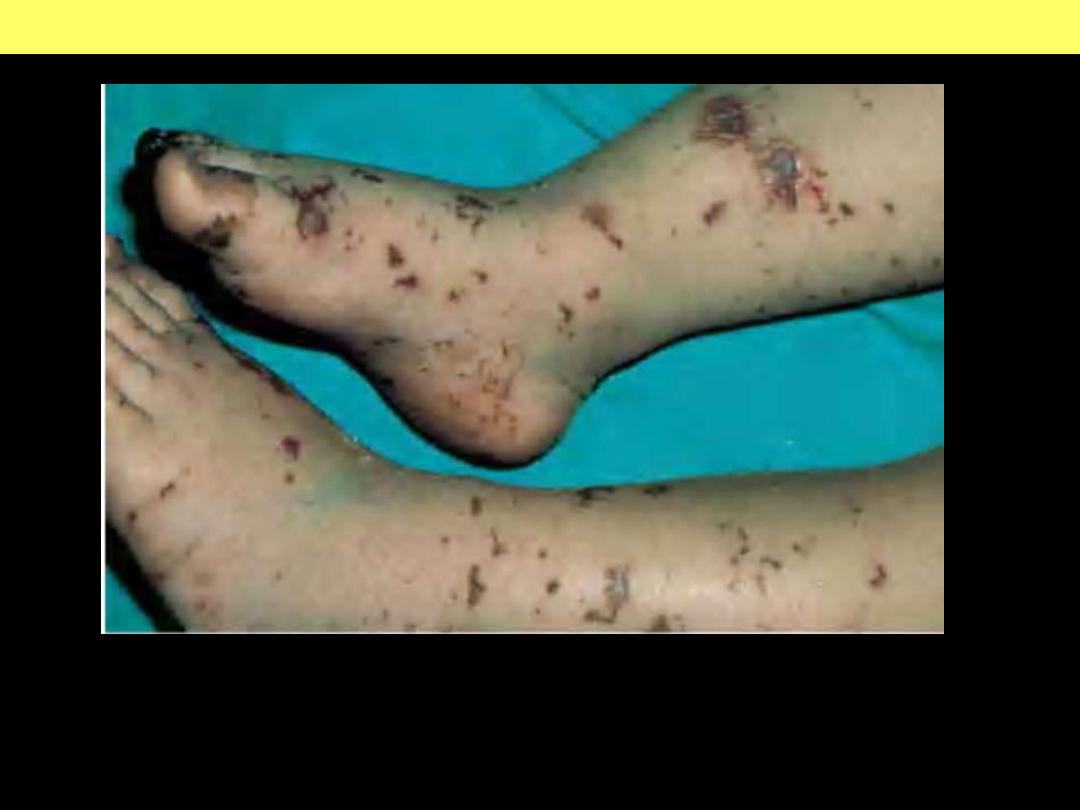
Purpura and necrosis with hemorrhagic bullae formation. Characteristic of
various
causes
of
disseminated
intravascular
coagulation
caused
by
microorganisms.
Meningococcemia: stellate purpura
Leukocytoclastic vasculitis is a small-vessel systemic vasculitis characterized by
the involvement of the skin as palpable purpura.
Leukocytoclastic vasculitis secondary to furosemide.
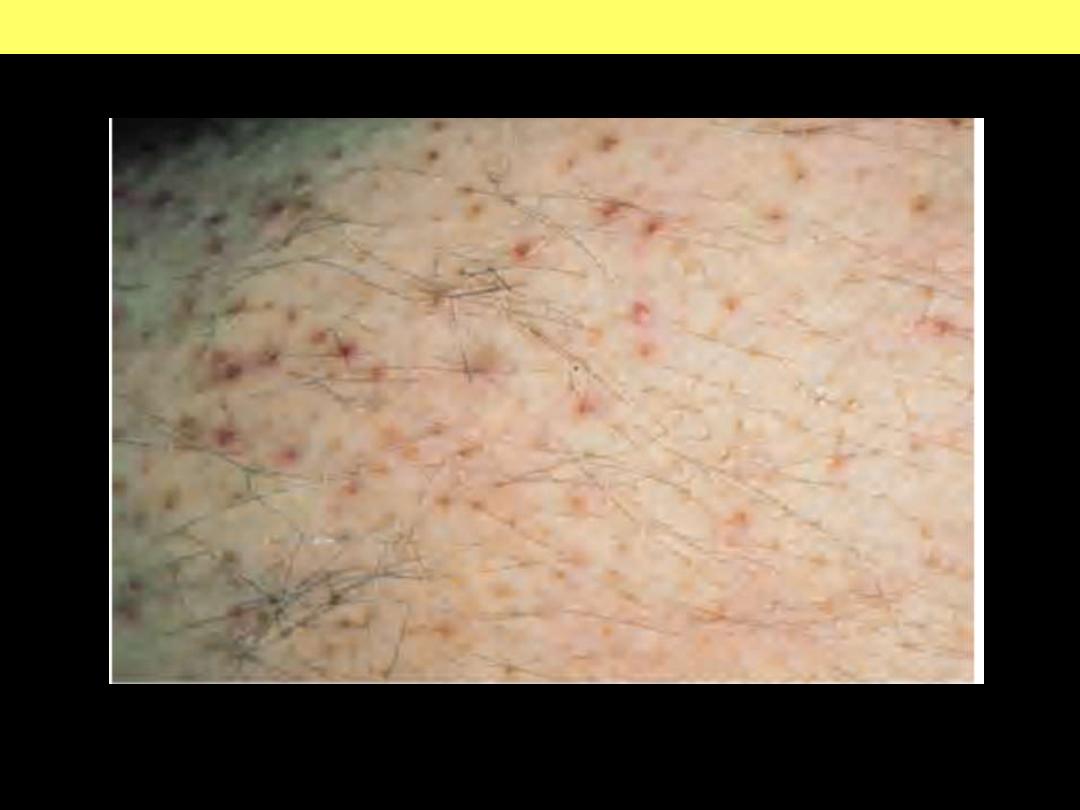
Scurvy. Vitamin C deficiency. Note parafollicular petechiae.
Scurvy
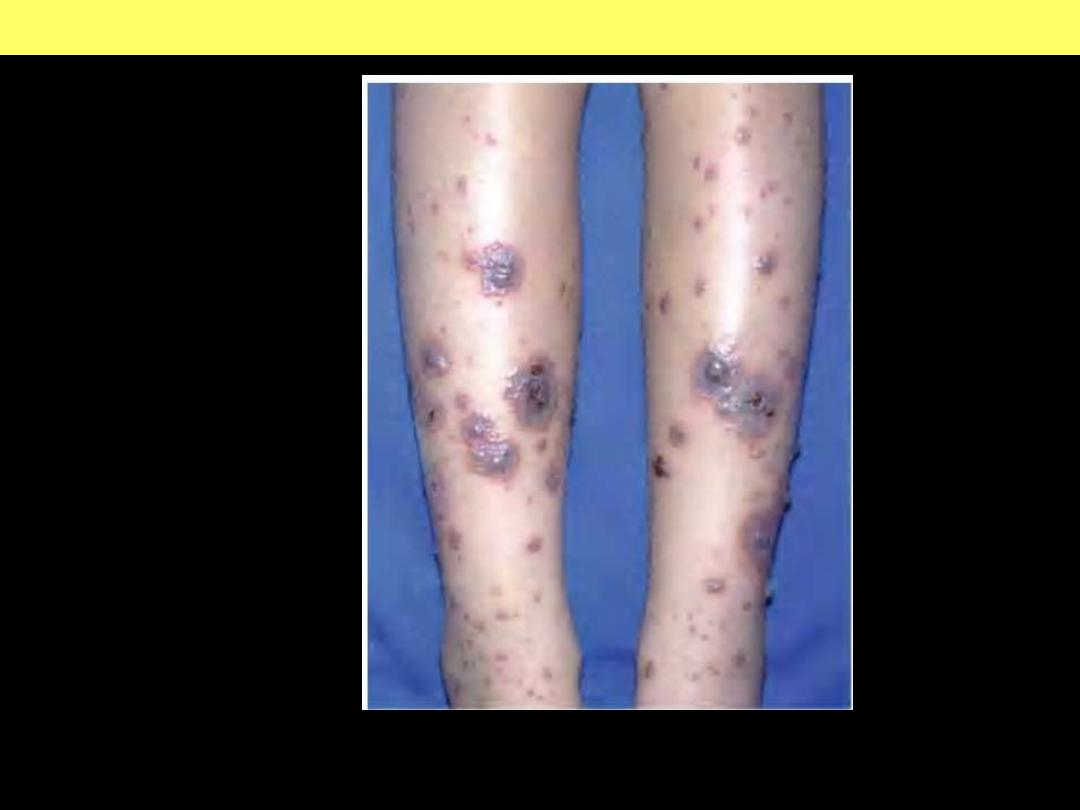
Urticarial papules and plaques can evolve into palpable non-thrombocytopenic
purpura.
Henoch-Schönlein purpura
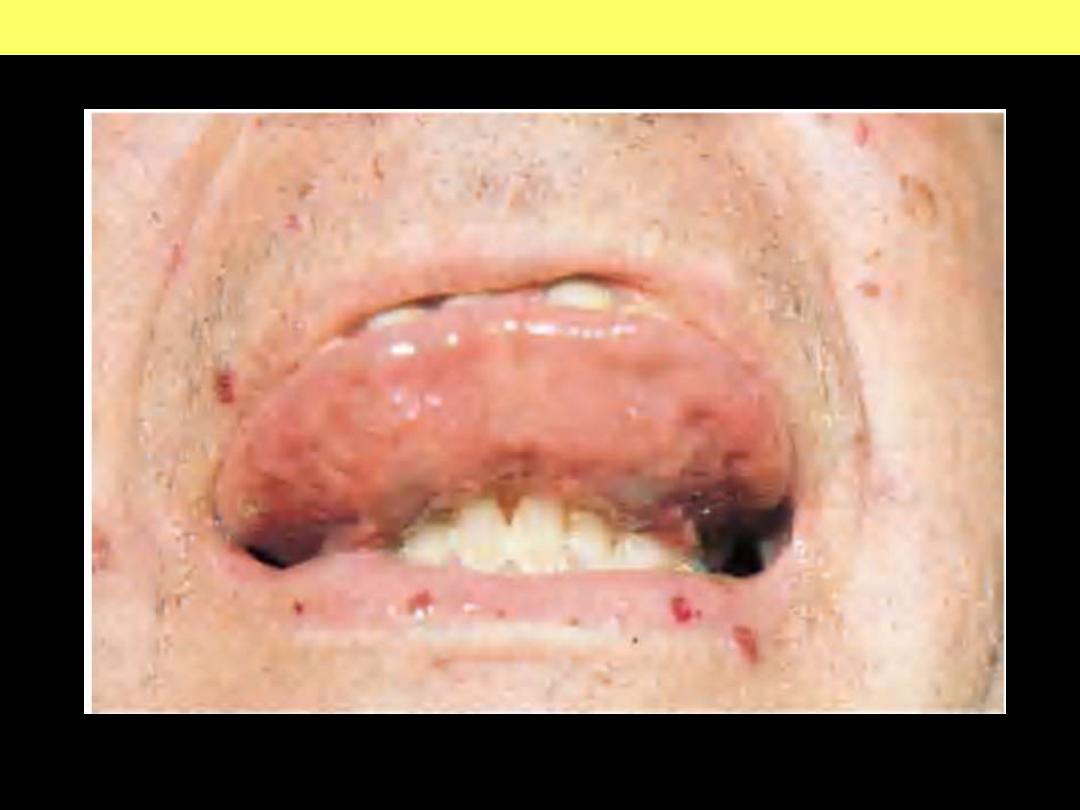
Sublingual and labial telangiectasia
Hereditary hemorrhagic telangiectasia

DIC
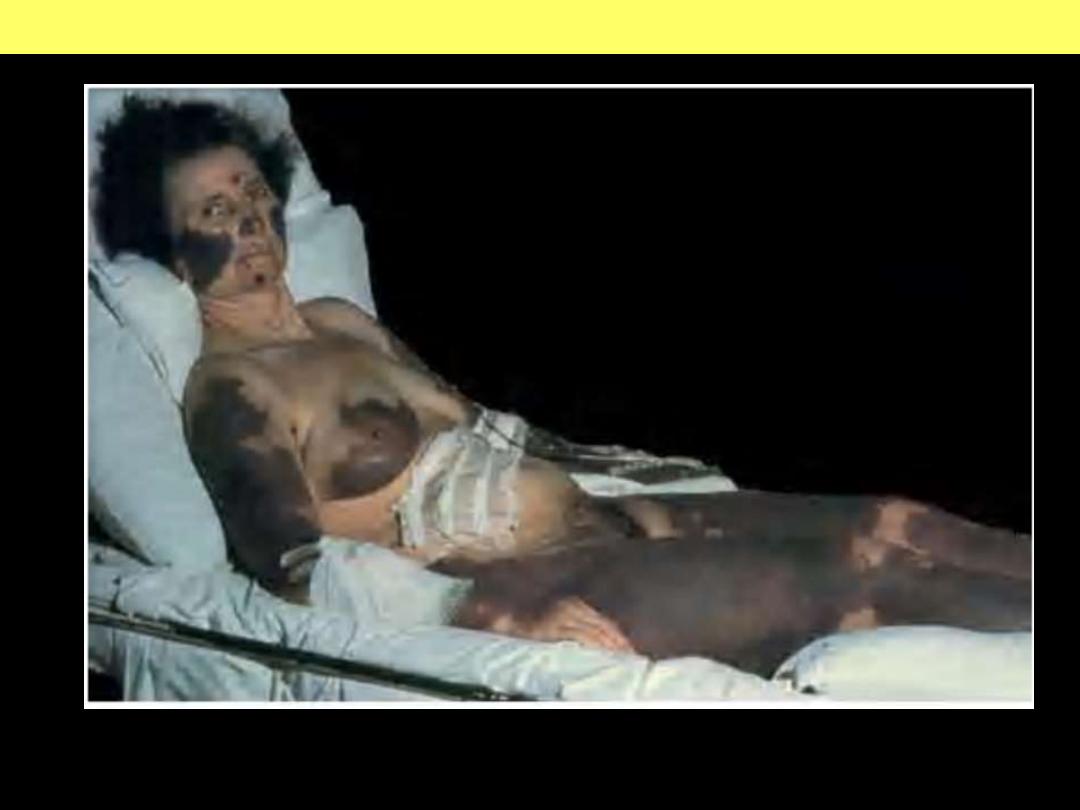
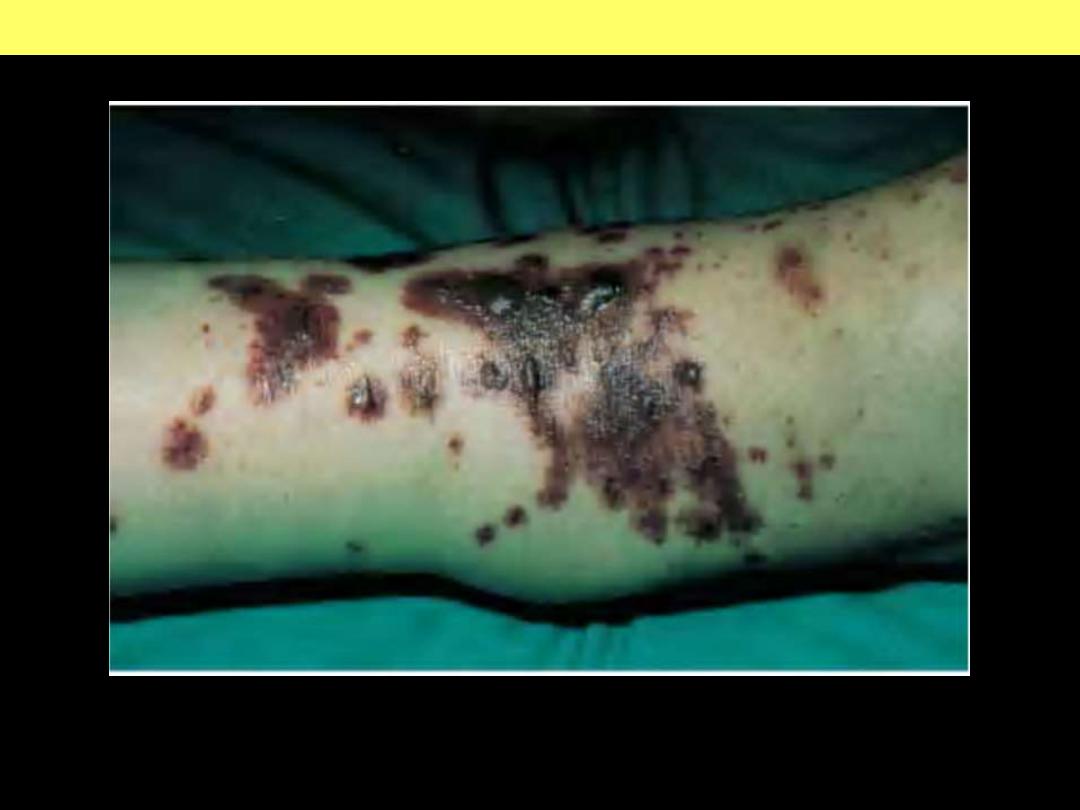
Extensive geographic areas of cutaneous infarction with hemorrhage involving the
face, breast, and extremities.
DISSEMINATED INTRAVASCULAR COAGULATION (DIC)
d
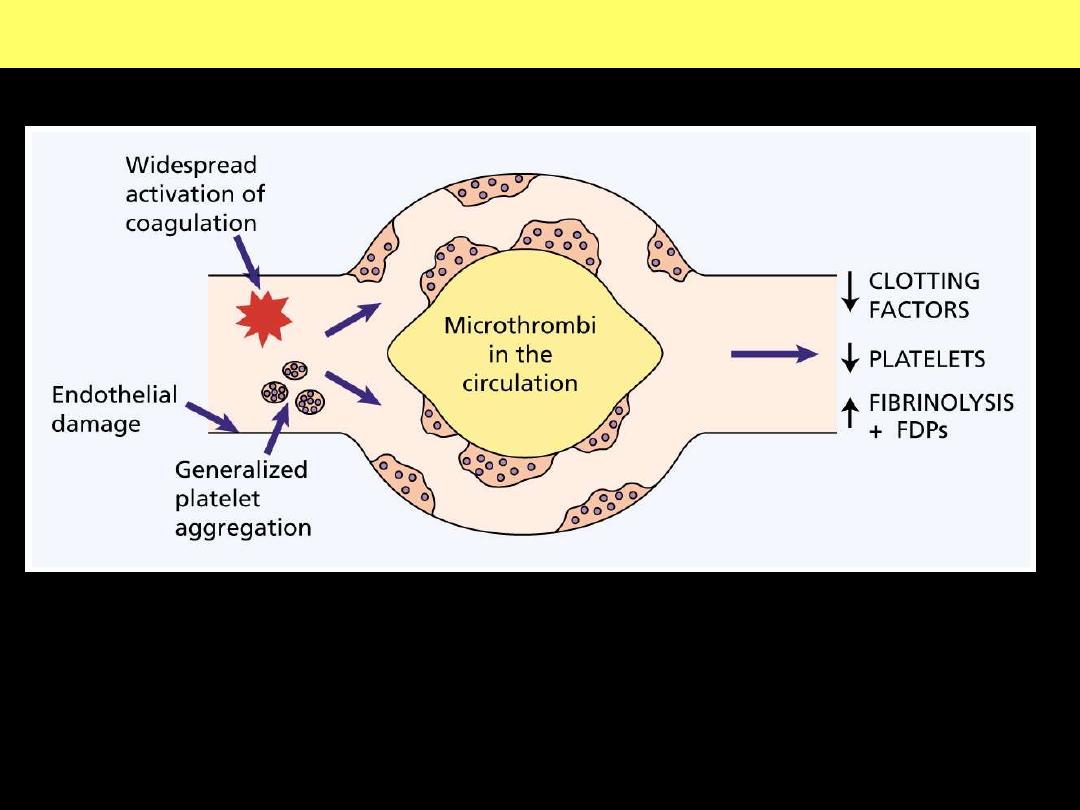
The pathogenesis of disseminated intravascular coagulation and the
changes in clotting factors, platelets and fibrin degradation products
(FDPs) that occur in this syndrome.
Pathogenesis of disseminated intravascular coagulation (DIC)
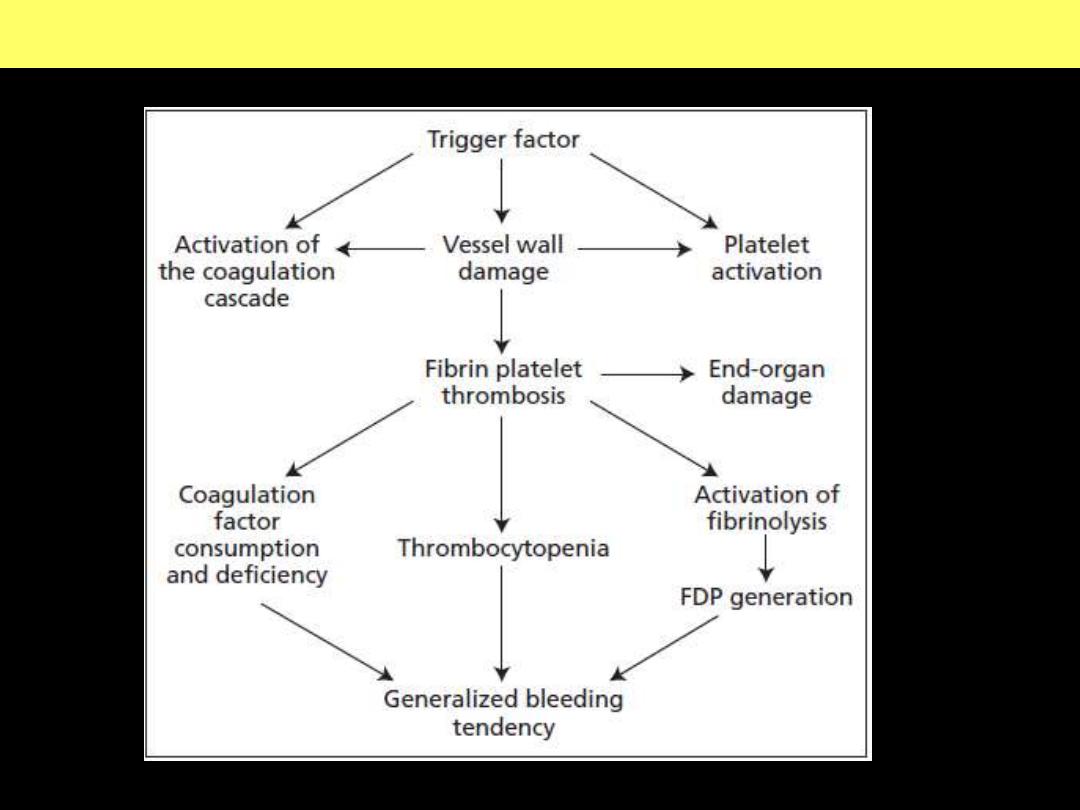
d
Pathogenesis of disseminated intravascular coagulation (DIC)

FactorVIII-vWF complex
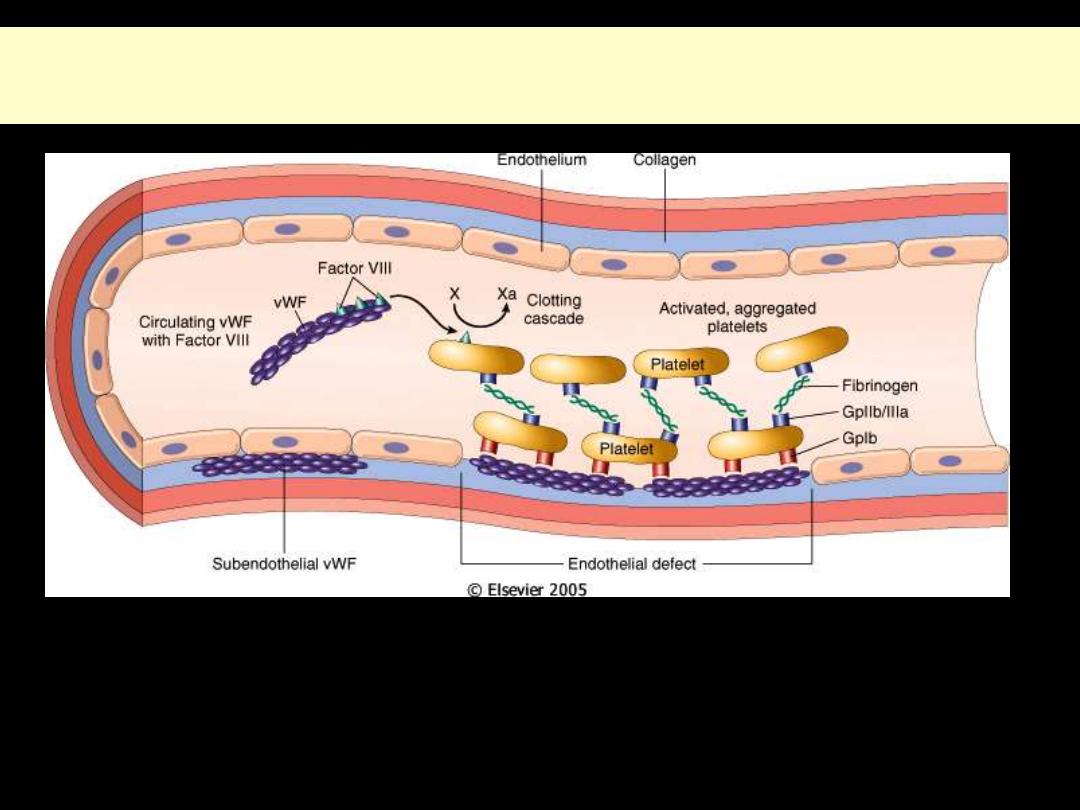
Structure and function of factor VIII-von Willebrand factor (vWF)
complex
Factor VIII is synthesized in the liver and kidney, and vWF is made in endothelial cells and
megakaryocytes. The two associate to form a complex in the circulation. vWF is also present in the
subendothelial matrix of normal blood vessels and the alpha granules of platelets. Following
endothelial injury, exposure of subendothelial vWF causes adhesion of platelets, primarily via
glycoprotein lb platelet receptor.
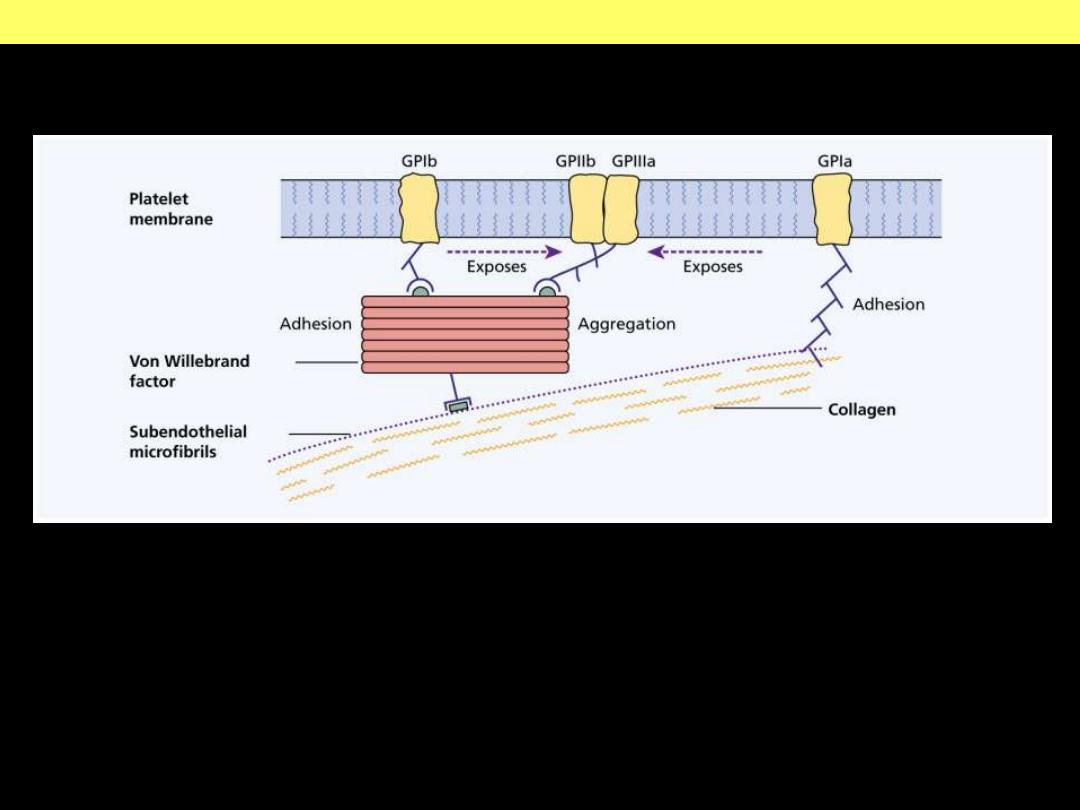
d
Structure and function of factor VIII-von Willebrand factor (vWF) complex

Hemophilia A - hemarthrosis
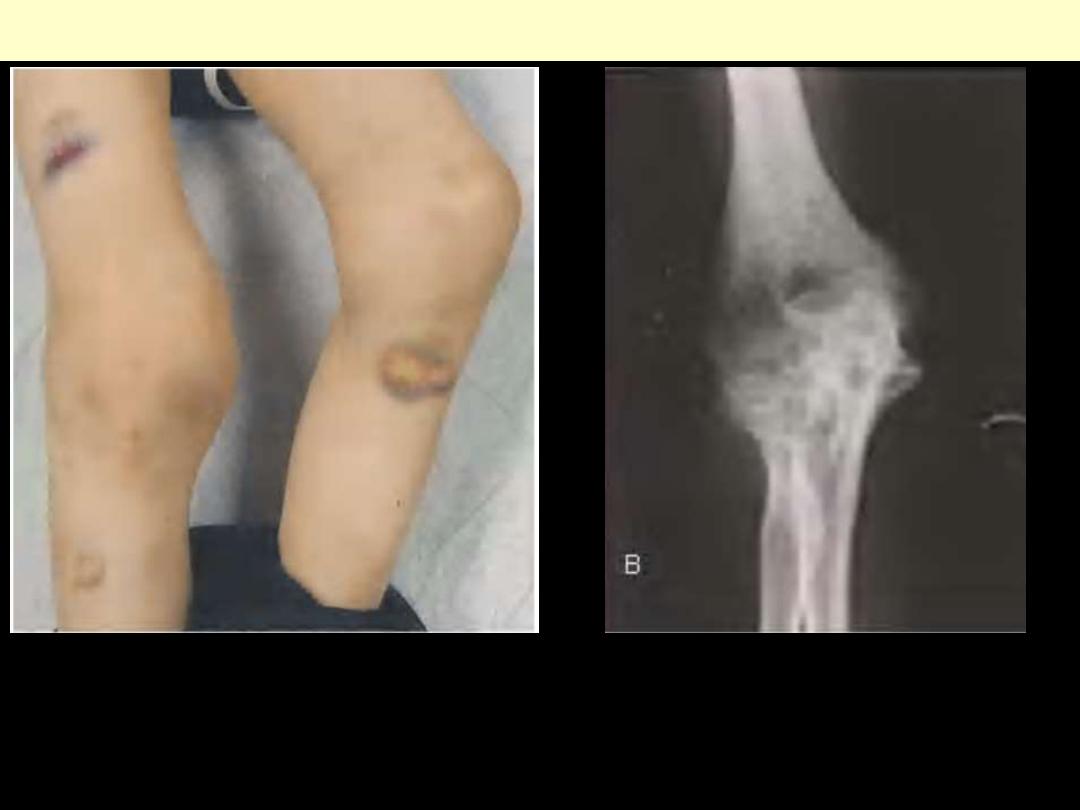
Hemophilia A - hemarthrosis
Hemophilia A: Hemarthrosis
Chronic right knee hemarthrosis with fresh and fading ecchymoses on legs.
Radiological image of knee showing loss of joint space with apparent fusion of
femoral and tibial articulation and cystic changes.

ITP pathogenesis diagram
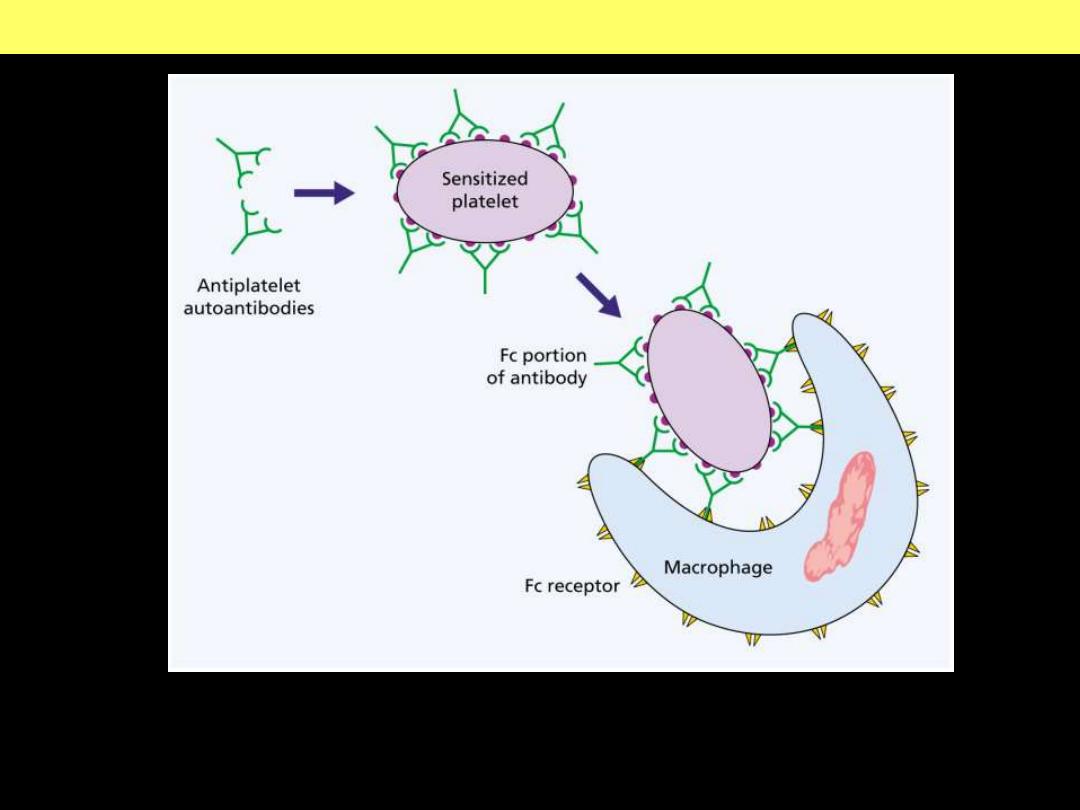
Pathogenesis of ITP
Chronic ITP is caused by the formation of
autoantibodies against platelet
membrane glycoproteins.
In the majority of cases, the antiplatelet antibodies are
of the IgG class.

Purpura - therombocytopenia
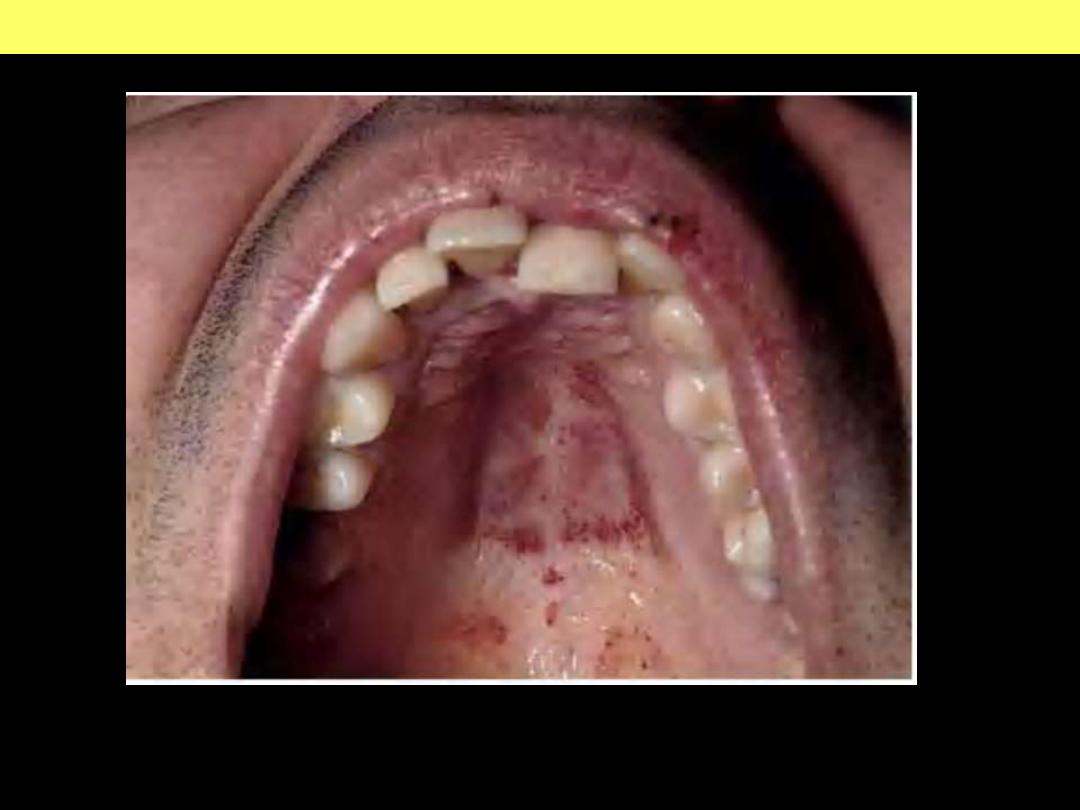
Thrombocytopenic purpura can first manifest on the oral mucosa or conjunctiva.
Here multiple petechial hemorrhages are seen on the palate.
Thrombocytopenic purpura

Thrombocytopenia – Drug-
induced mechanism
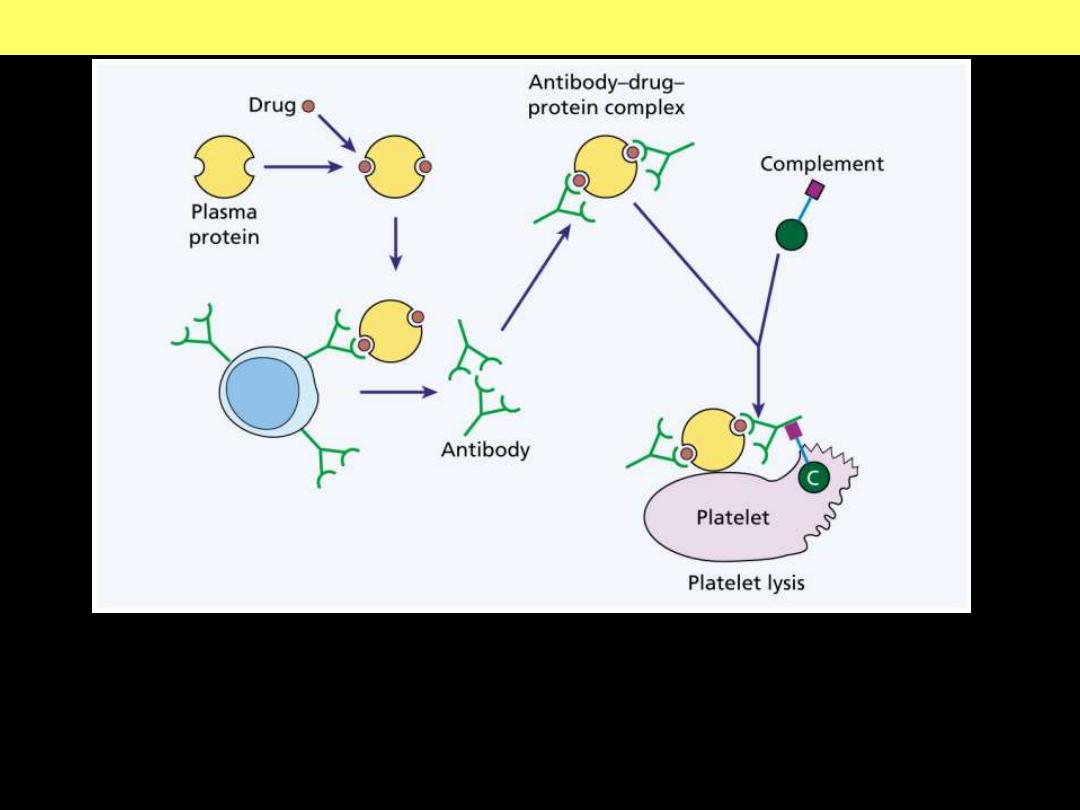
Mechanisms of drug induced thrombocytopenia
An antibody-drug-protein complex is deposited on the platelet surface. If
complement is attached and the sequence goes to completion, the platelet may be
lysed directly. Otherwise, it is removed by reticuloendothelial cells because of
opsonization with immunoglobulin and / or the C3 component of complement.

RBC disorders
F:\lectures\4th
grade\Pathology\New folder

Aplastic anemia
F:\lectures\4th
grade\Pathology\New folder
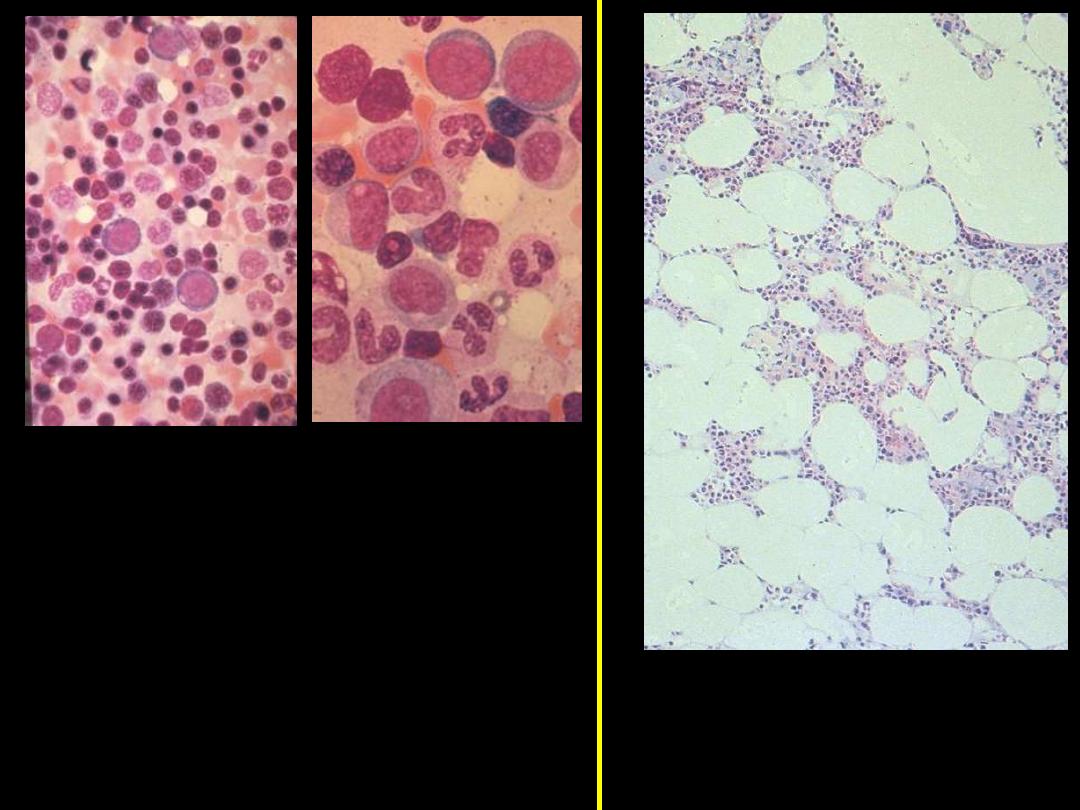
Bone marrow aspirate: shows
myeloid precursors ranging from
myeloblasts to segmented
neutrophils. Several erythroid
precursors with condensed
nuclear chromatin are also seen.
This specimen from a bone
marrow aspirate is very
hypocellular.
Normal
Aplastic Anemia

Autoimmune hemolytic anemia
F:\lectures\4th
grade\Pathology\New folder
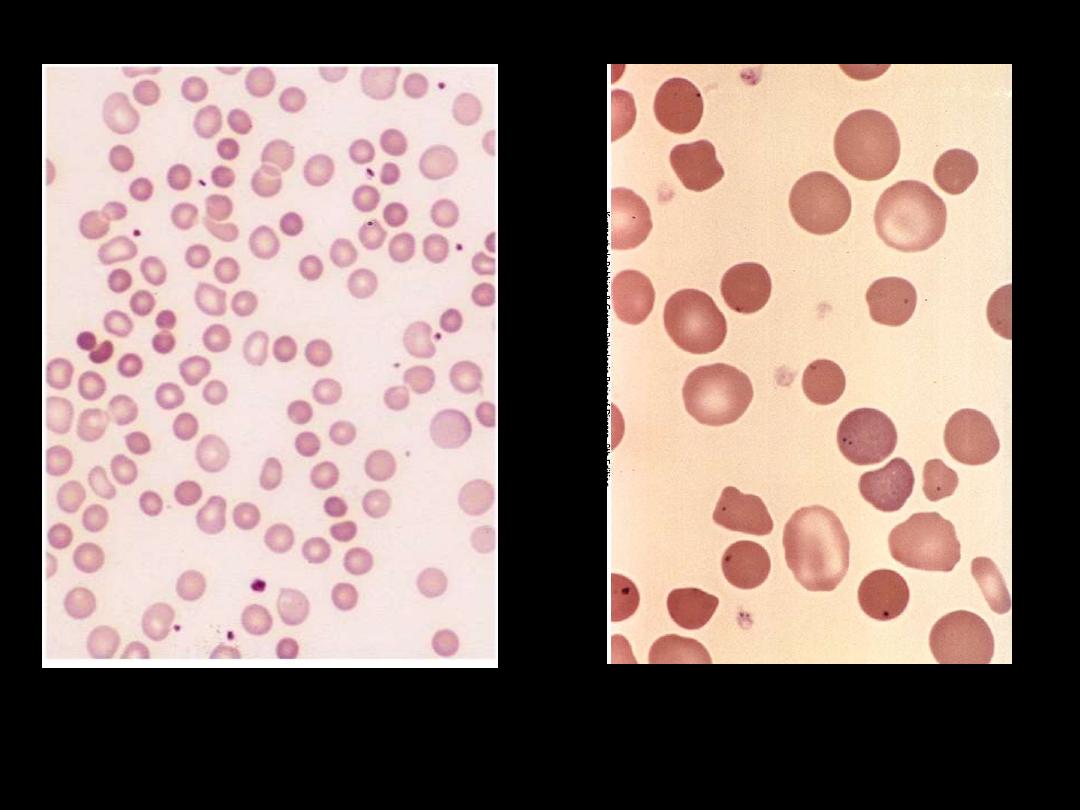
The blood film shows microspherocytes which are densely
staining with smaller diameters than normal red cells.
Warm type Autoimmune hemolytic anemia
Red cell agglutination. Several clumps of agglutinated
red cells, two are marked by arrows.
Cold type Autoimmune hemolytic anemia

G6PD deficiency
F:\lectures\4th
grade\Pathology\New folder
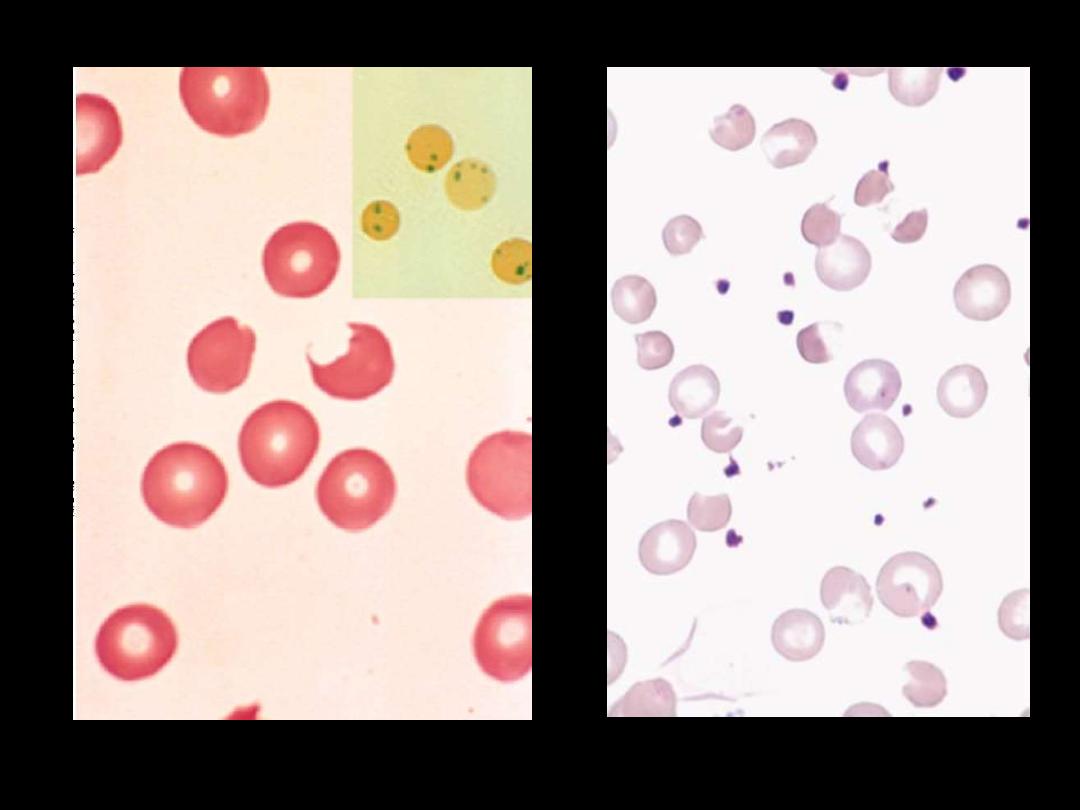
The blood film may show
contracted and fragmented cells, 'bite' cells and
'blister‘ cells
which have had Heinz bodies removed by the spleen.
GIucose-6-phosphate dehydrogenase deficiency
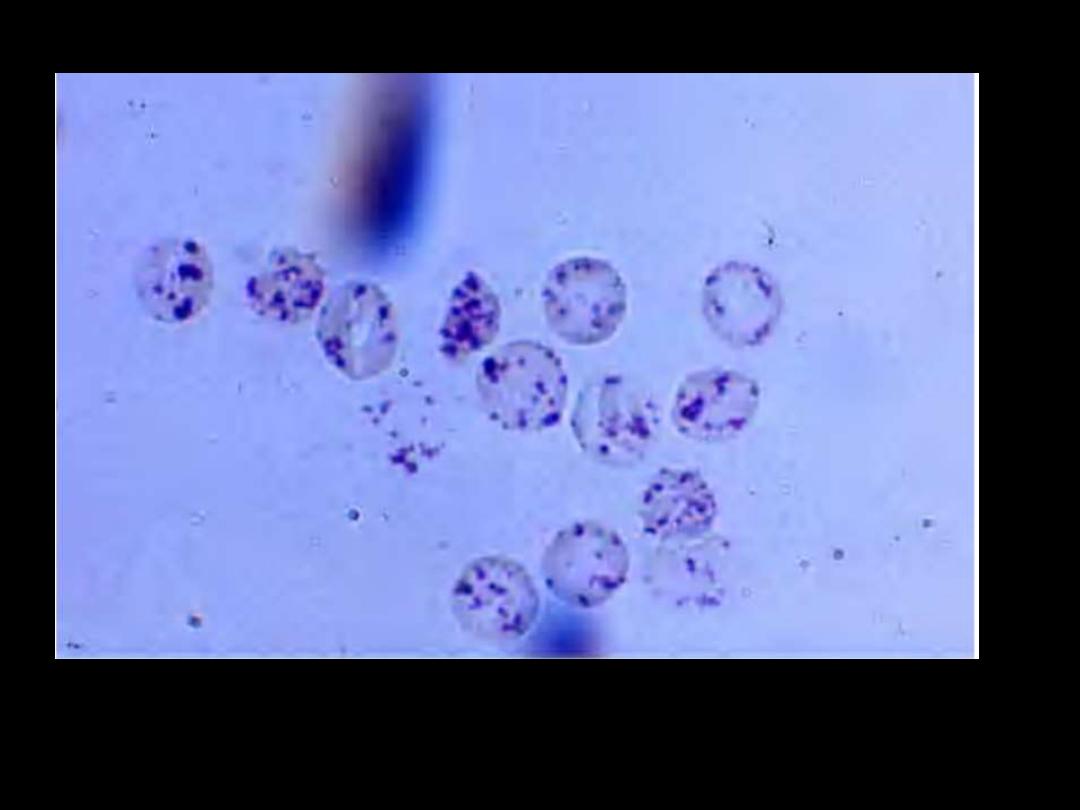
Heinz bodies: Supravital stain. These bodies are particles of
denatured hemoglobin, usually attached to the inner face of the red
cell membrane.
Heinz bodies: G6PD deficiency
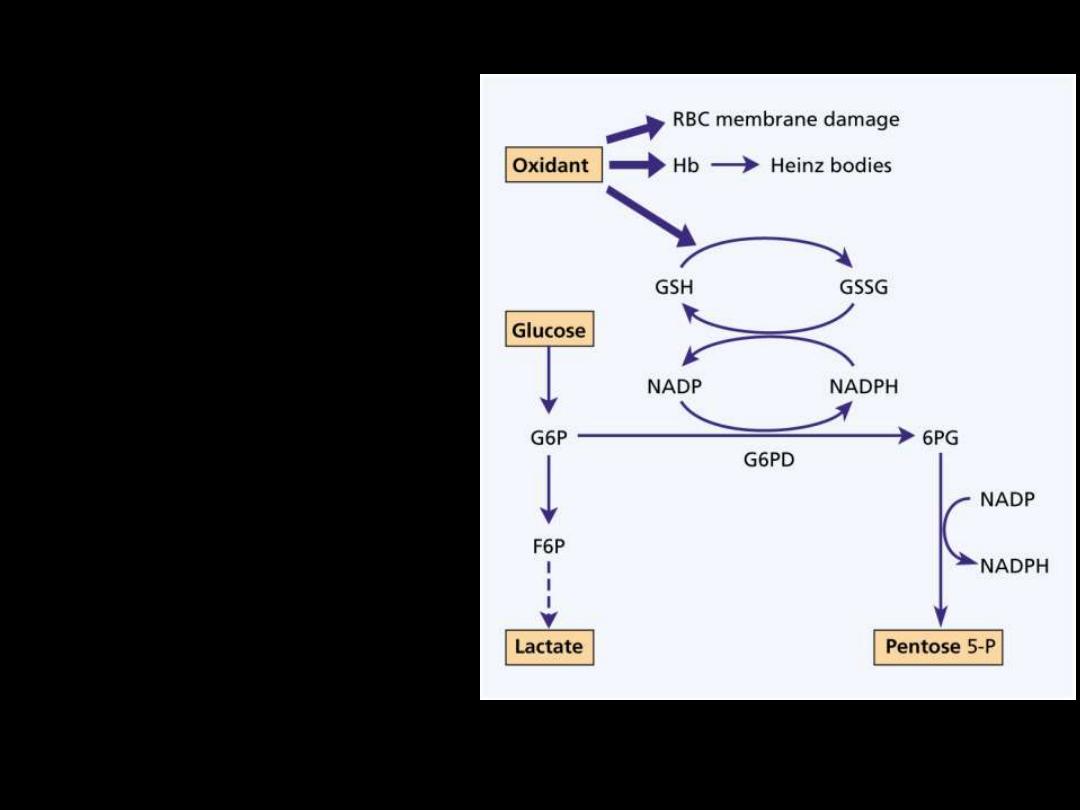
GIucose-6-phosphate
dehydrogenase deficiency:
Glucose-6-phosphate
dehydrogenase (G6PD)
functions to reduce
nicotinamide adenine
dinucleotide phosphate
(NADP) while oxidizing
glucose-6-phosphate.
It is the only source of
NADP in red cells
.
As NADP is needed for the
production of reduced
glutathione a deficiency
renders the red cell
susceptible to oxidant
stress.

Hereditary spherocytosis
F:\lectures\4th
grade\Pathology\New folder
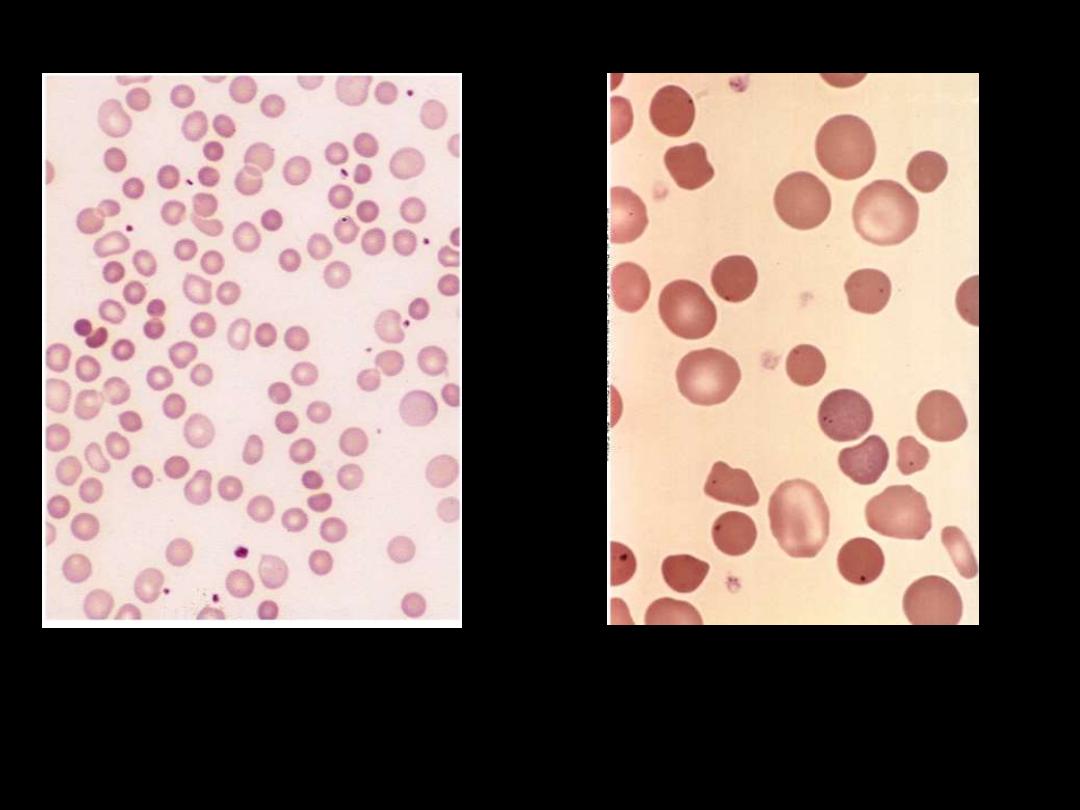
Note the anisocytosis and several dark-appearing spherocytes with
no central pallor. Howell-Jolly bodies (small dark nuclear
remnants) are also present in red cells of this asplenic patient.
Hereditary spherocytosis
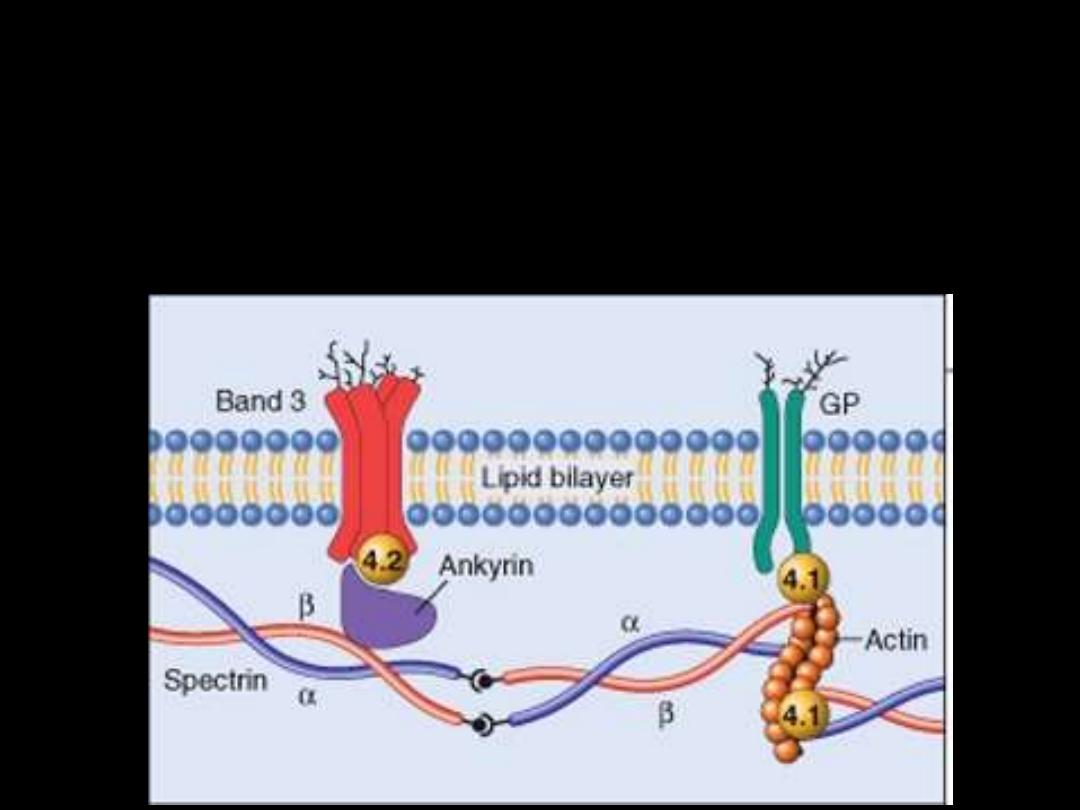
Pathogenesis:
HS is usually caused by defects in:
The proteins involved in the vertical interactions between the
membrane skeleton and the lipid bilayer of the red cell.
Various mutations involving:
Spectrin and Ankyrin that weaken the interactions between these
proteins cause red cells to lose membrane fragments.
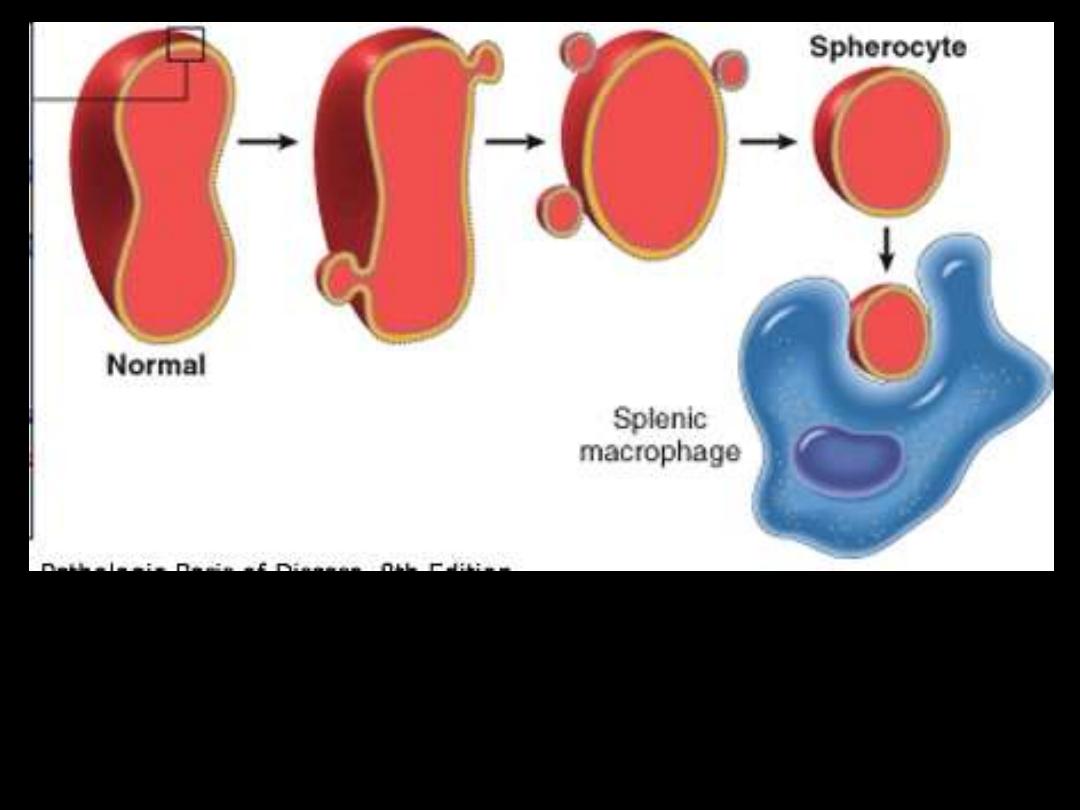
The loss of membrane may be caused by the
release of parts of the lipidbilayer that are not
supported by the skeleton.

Iron deficiency anemia
F:\lectures\4th
grade\Pathology\New folder
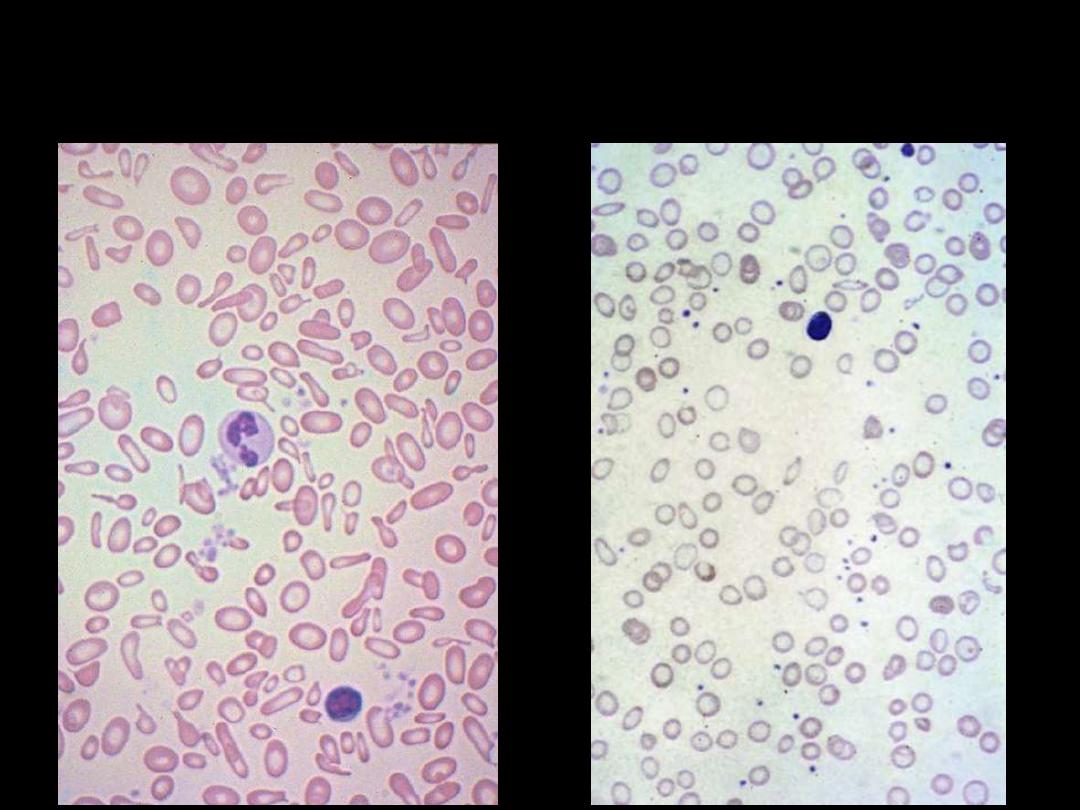
Iron Deficiency Anemia: Blood Film
The blood film
shows
hypochromic microcytic
cells with
occasional
pencil-shaped poikilocytes.

Megaloblastic anemia
F:\lectures\4th
grade\Pathology\New folder
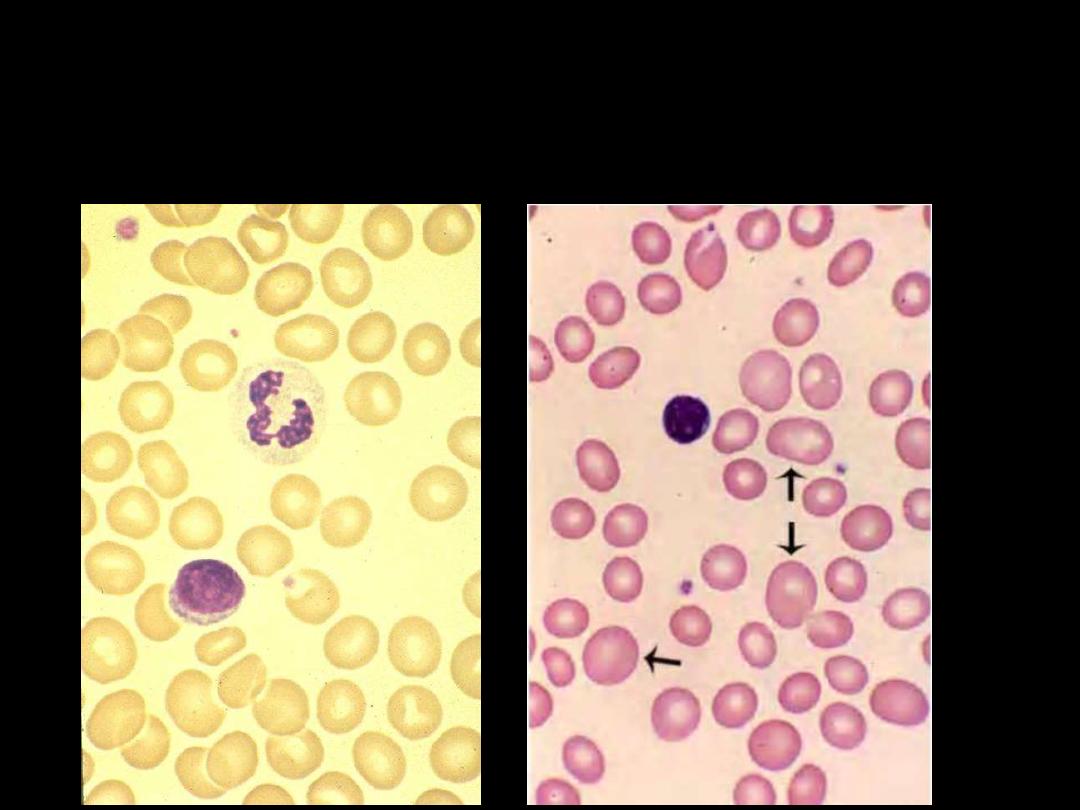
Megaloblastic Anemia:
The anemia is macrocytic (MCV >95 fL). The
macrocytes are typically oval in shape. The reticulocyte count is low. The
total white cell and platelet counts may be moderately reduced,
especially in severely anaemic patients.
Normal
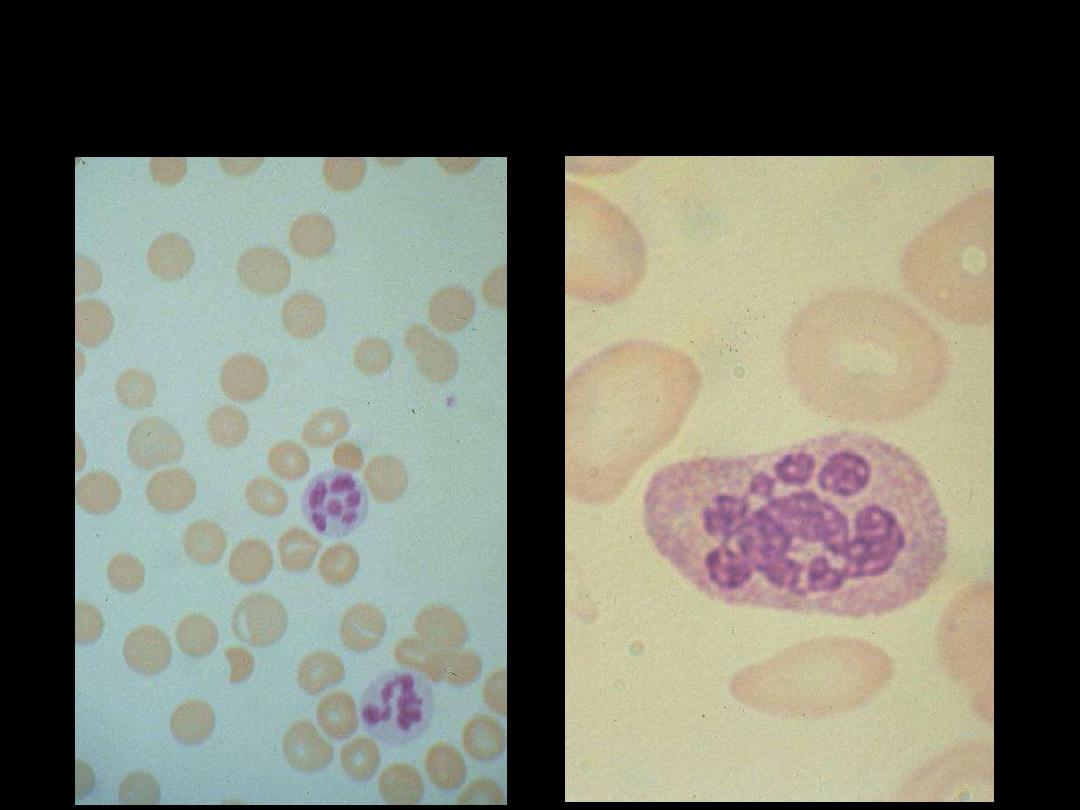
Megaloblastic Anemia:
A proportion of the neutrophils
show
hypersegmented nuclei
(with six or more lobes)
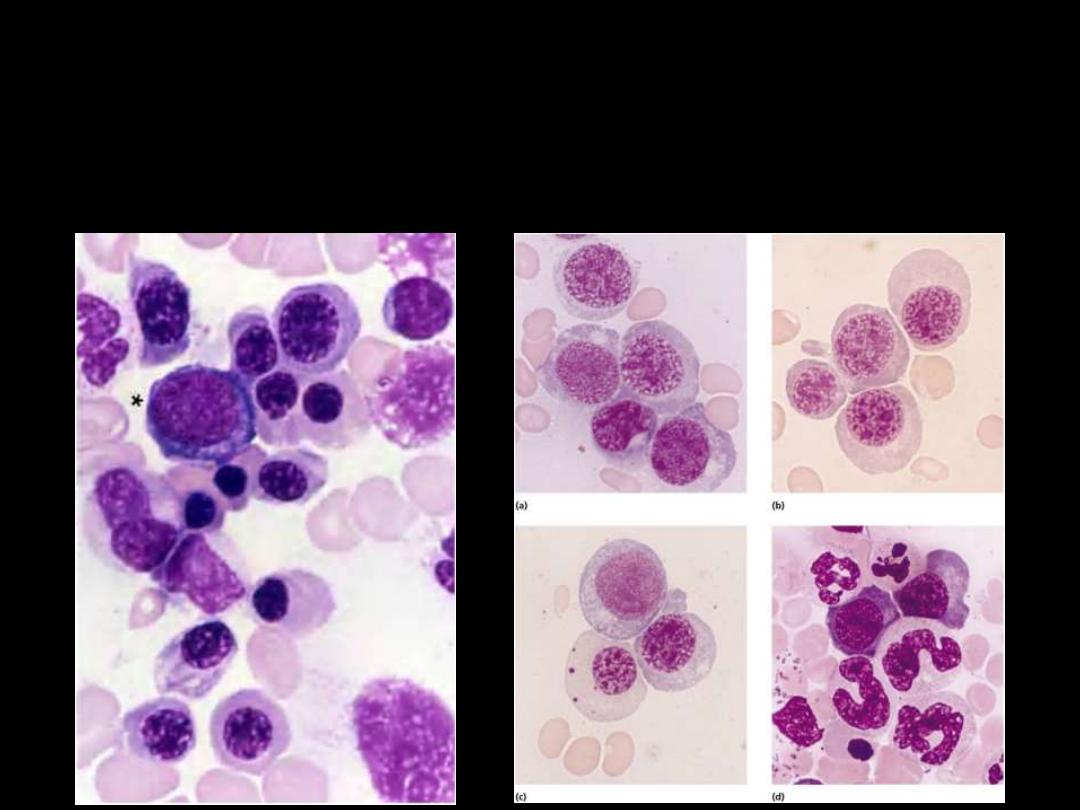
Megaloblastic Anemias
: The bone marrow
is usually hypercellular
and The erythroblasts are large and show failure of nuclear
maturation maintaining an open, fine, lacy primitive chromatin
pattern but normal hemoglobinization
Normal

Microangiopathic hemolytic
anemia
F:\lectures\4th
grade\Pathology\New folder
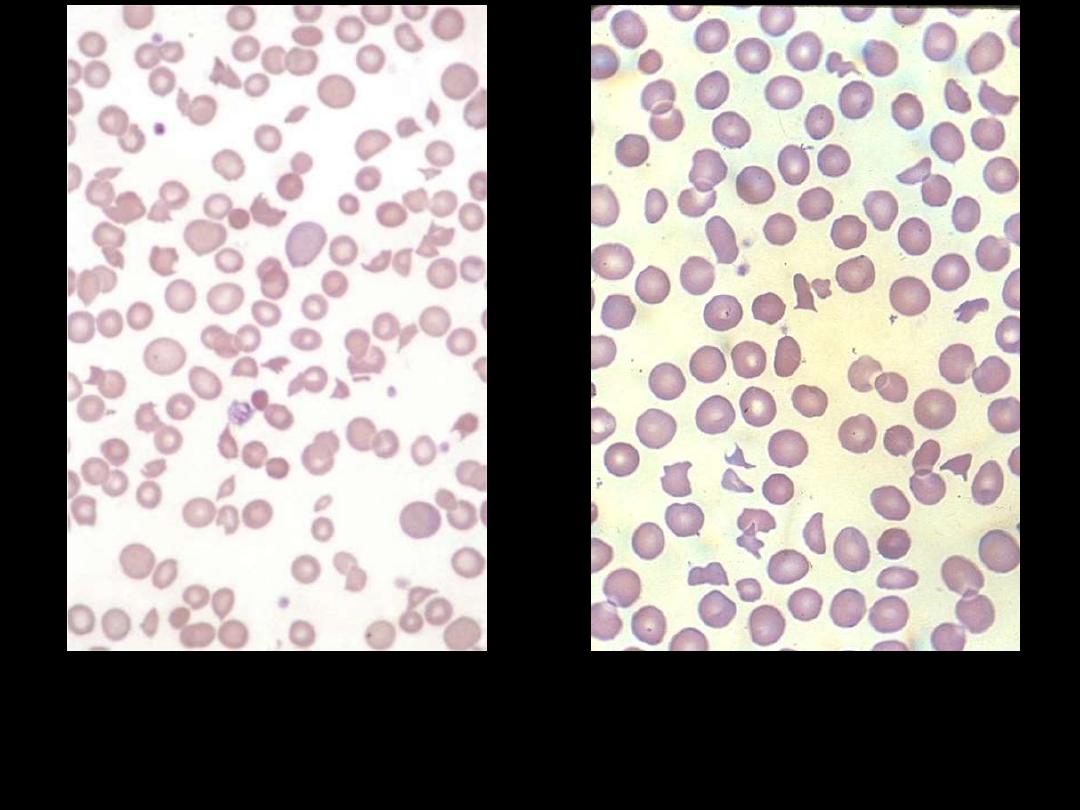
Blood film in microangiopathic hemolytic anemia. Numerous
contracted and deeply staining cells and cell fragments are
present.

Sickle-cell anemia
F:\lectures\4th
grade\Pathology\New folder
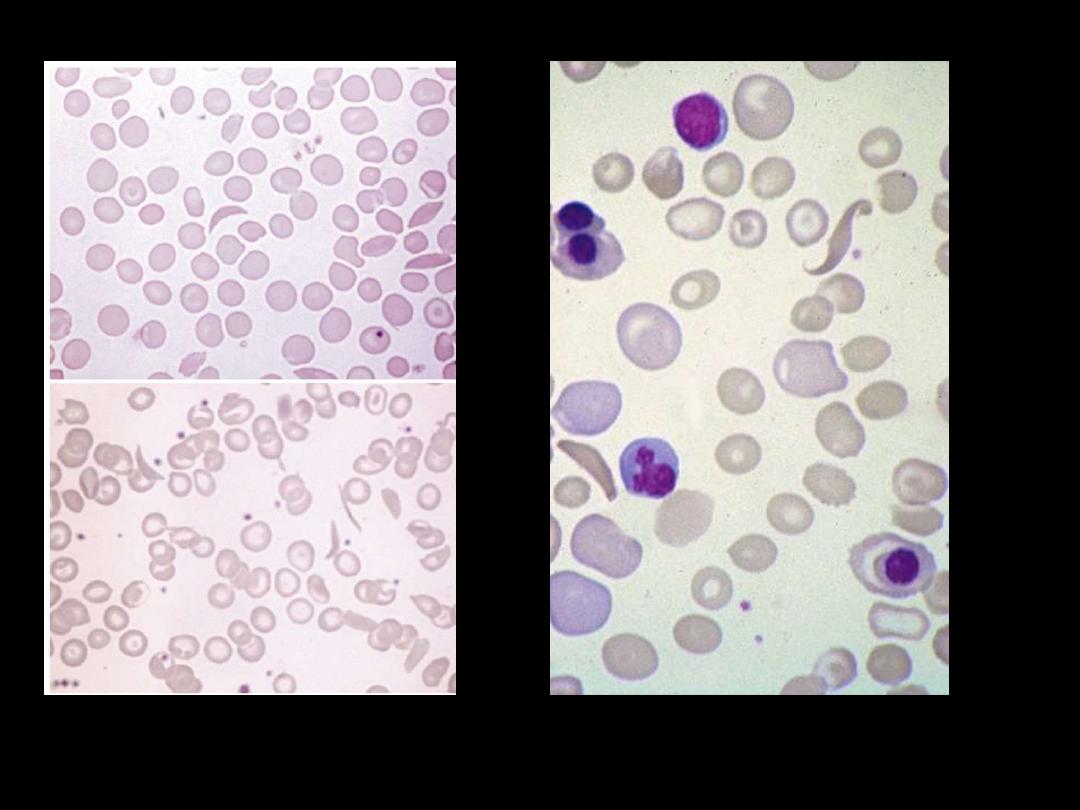
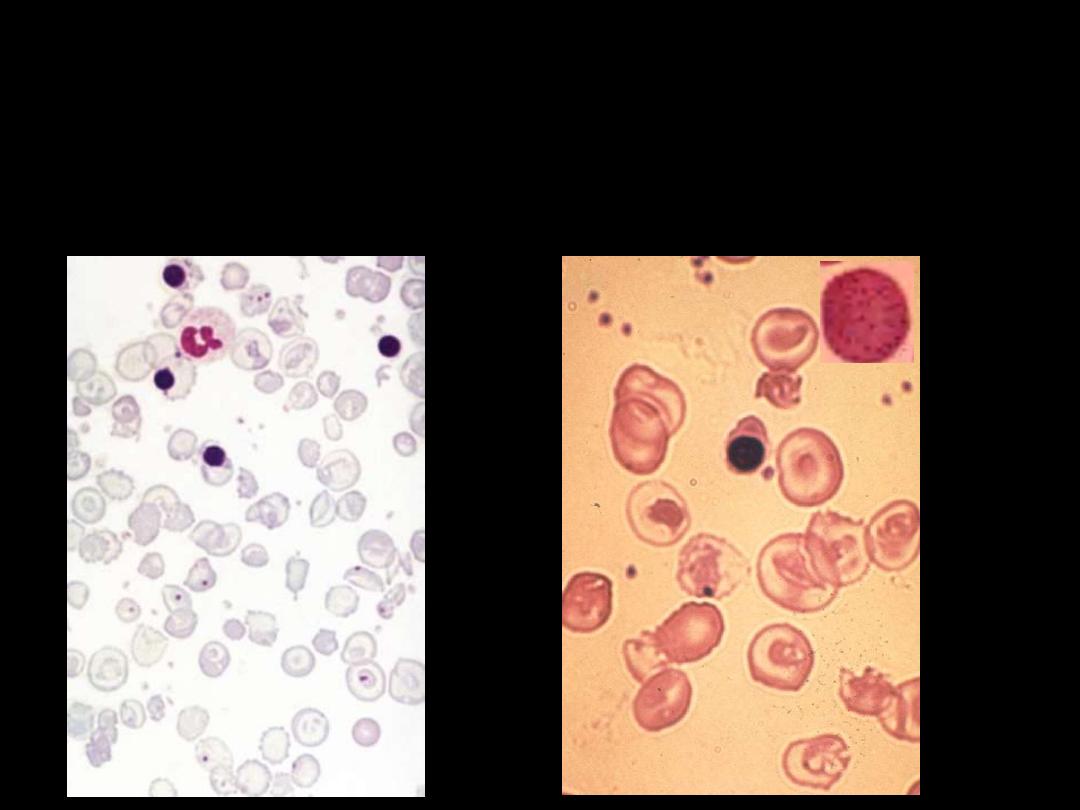
Sickle cell anaemia: peripheral blood film showing deeply staining
sickle cells, target cells and polychromasia.
Sickle cell anemia
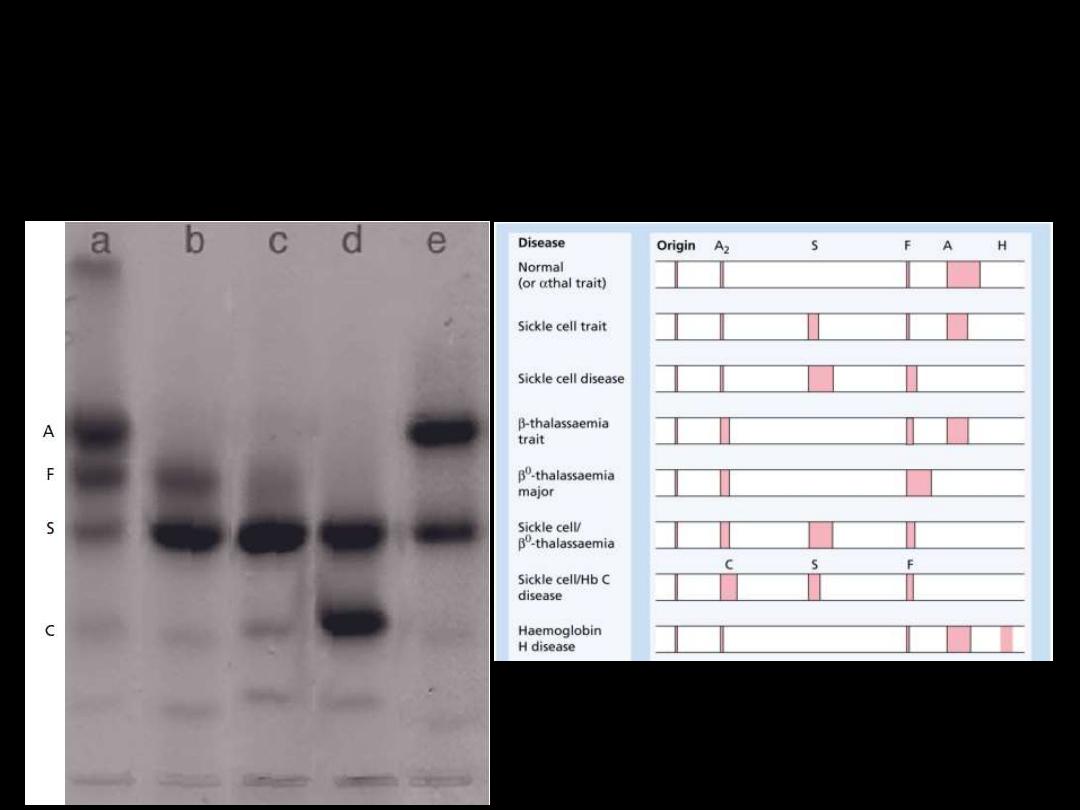
Sickle cell anemia
●Hemoglobin electrophoresis:
In Hb SS:
No Hb A is detected.
The amount of
Hb F
is variable and is usually
5-15%.
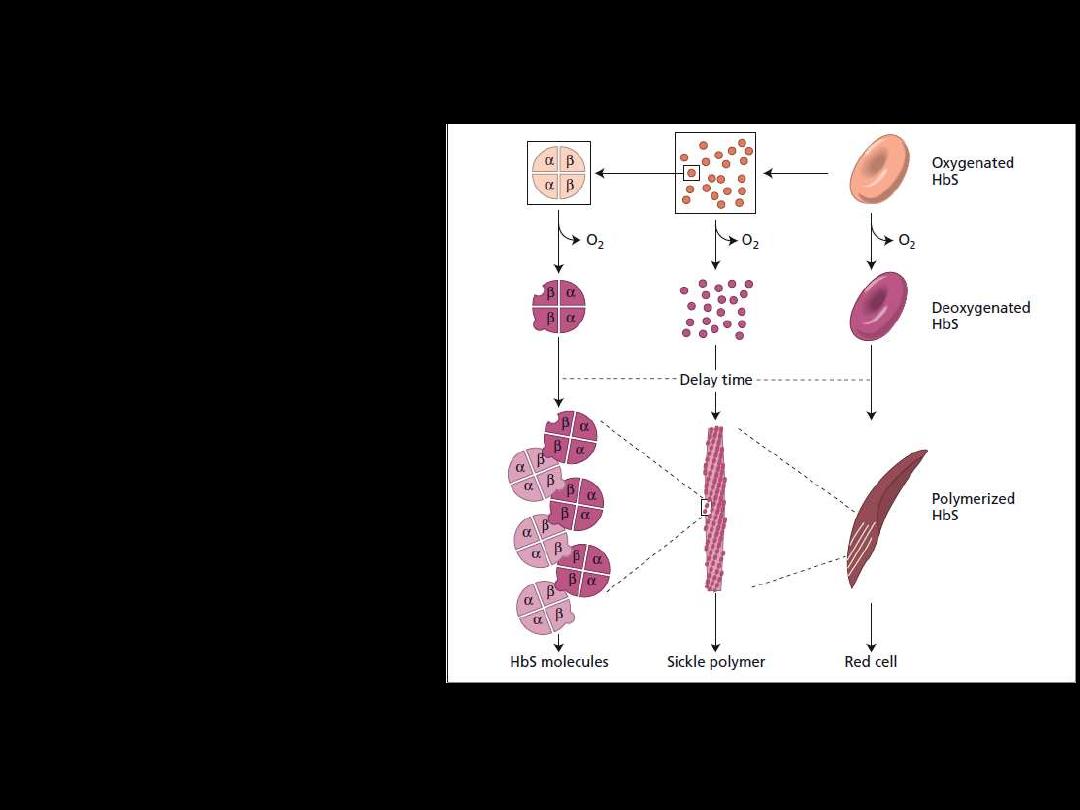
Pathogenesis:
Hb S (Hb α
2
β
2
S
) is insoluble
and forms crystals when
exposed to low oxygen
tension.
Deoxygenated sickle
hemoglobin polymerizes into
long fibres, each consisting of
seven intertwined double
strands with cross-linking.
The red cells sickle and may
block different areas of the
microcirculation or large
vessels causing infarcts of
various organs.
The sickle β-globin
abnormality is caused by
substitution of valine for
glutamic acid in position 6 in
the β chain
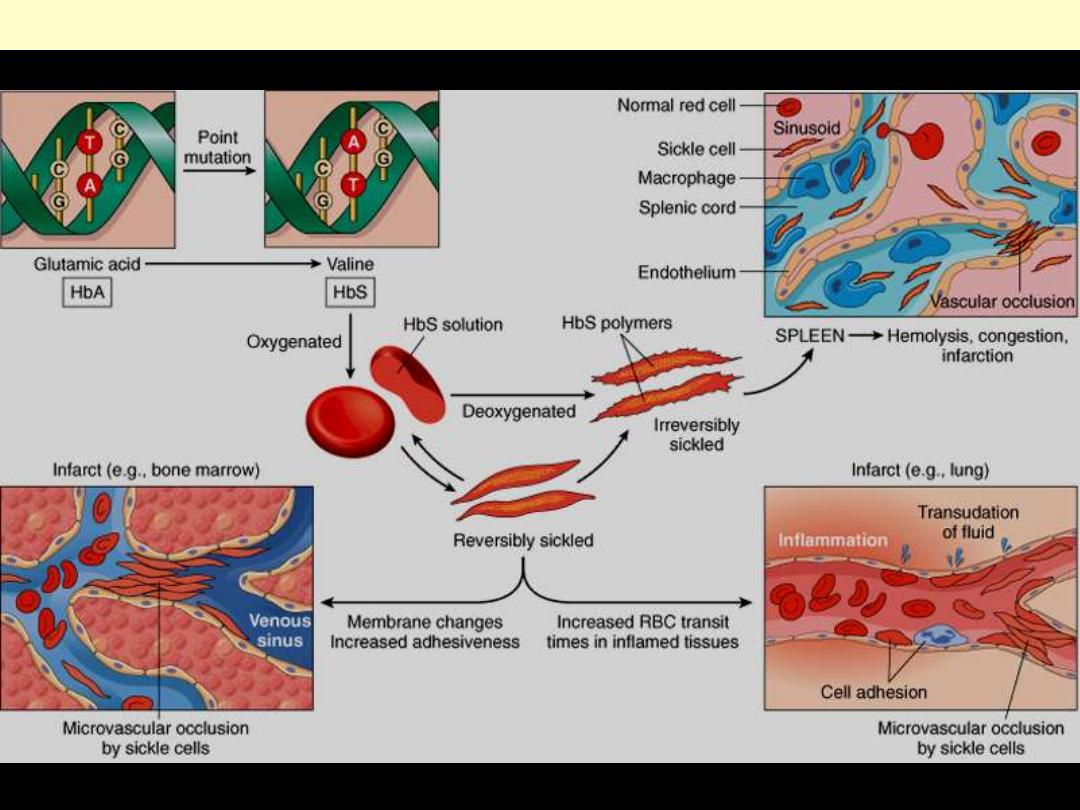
d
Pathophysiology and morphologic consequences of sickle cell anemia

Thalassemias
F:\lectures\4th
grade\Pathology\New folder
Laboratory diagnosis: (β-Thalassemia Major)
●There is a severe hypochromic, microcytic anemia.
●Raised reticulocyte percentage.
●Normoblasts, target cells and basophilic stippling in the blood
film.
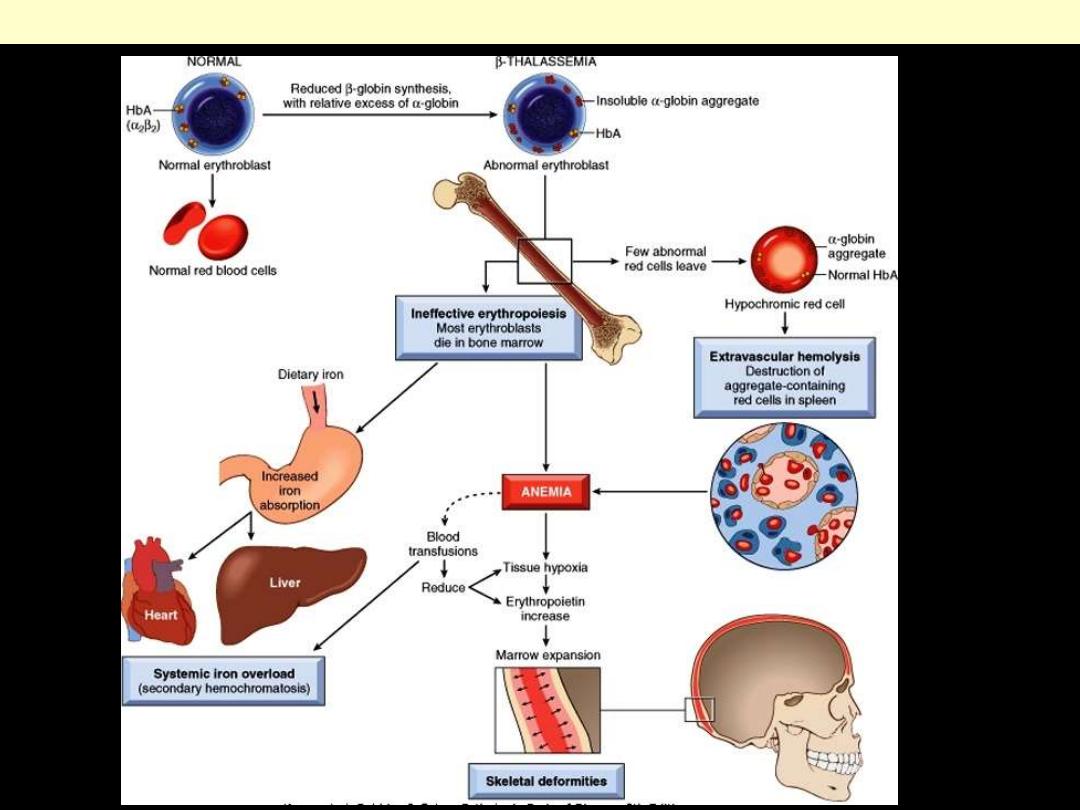
Pathogenesis of β-thalassemia major
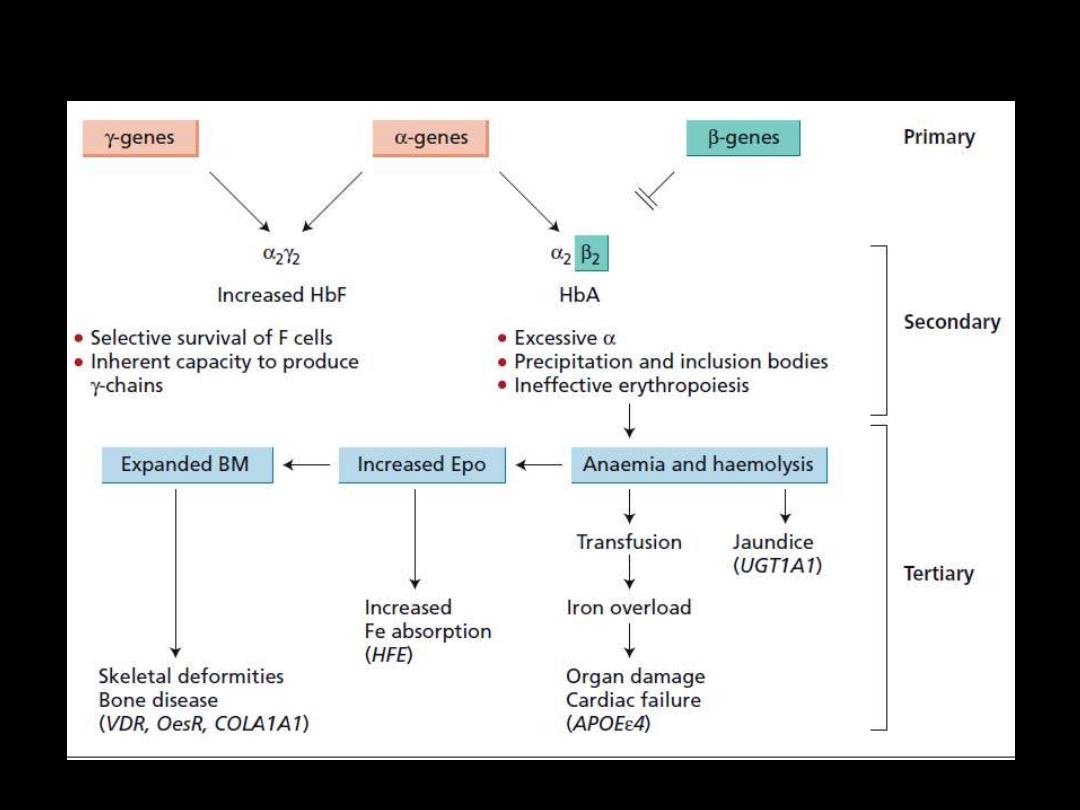
Pathophysiology: β-Thalassemia Major
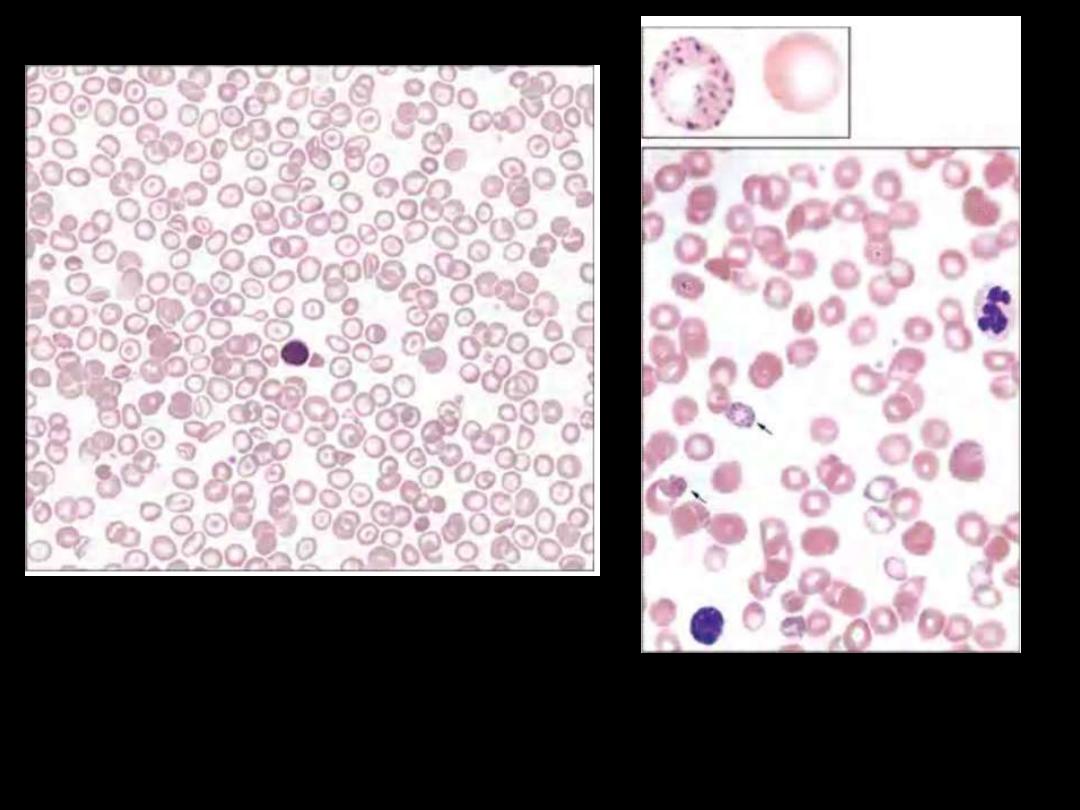
Heterozygous beta thalassemia.
Some variation in size and shape is
apparent, as is modest microcytosis
and hypochromia. Platelets are also
seen.
Basophilic stippling in
thalassemia.
β-Thalassemia Minor
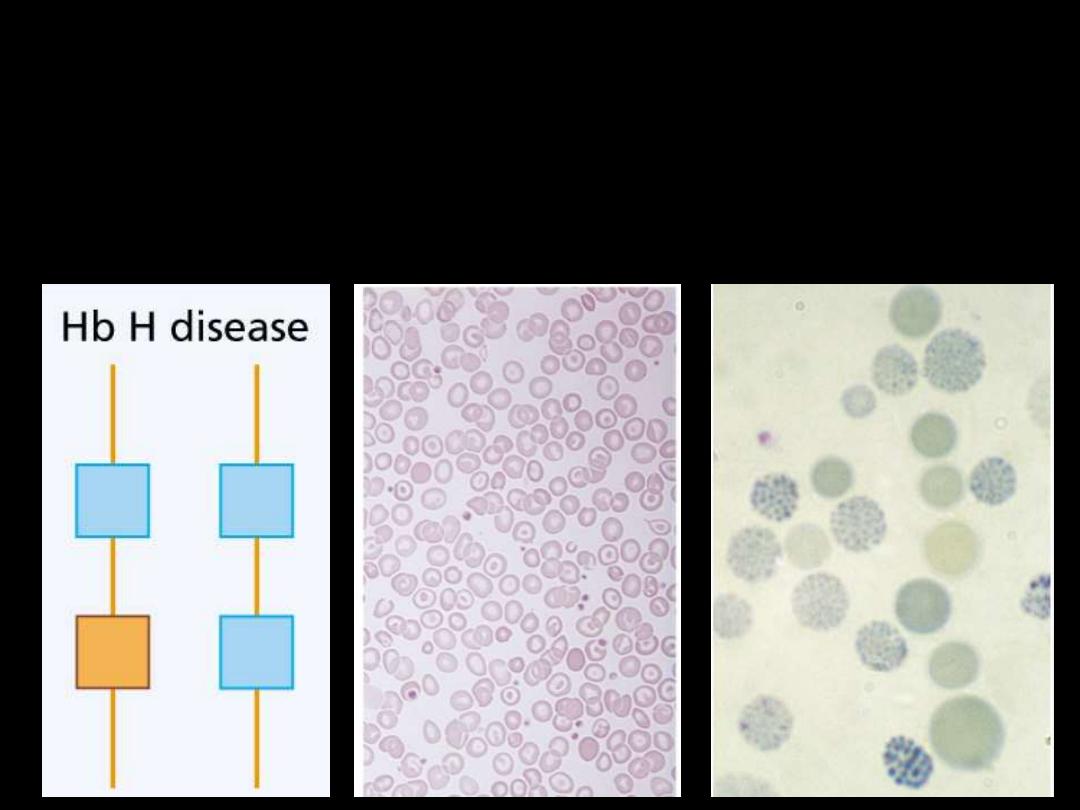
Hb H Disease:
Three α gene deletions
leads to a moderately severe
(hemoglobin 7-11 g/dL) microcytic, hypochromic anemia
with splenomegaly.

WBC disorders
F:\lectures\4th
grade\Pathology\New folder

AL
F:\lectures\4th
grade\Pathology\New folder
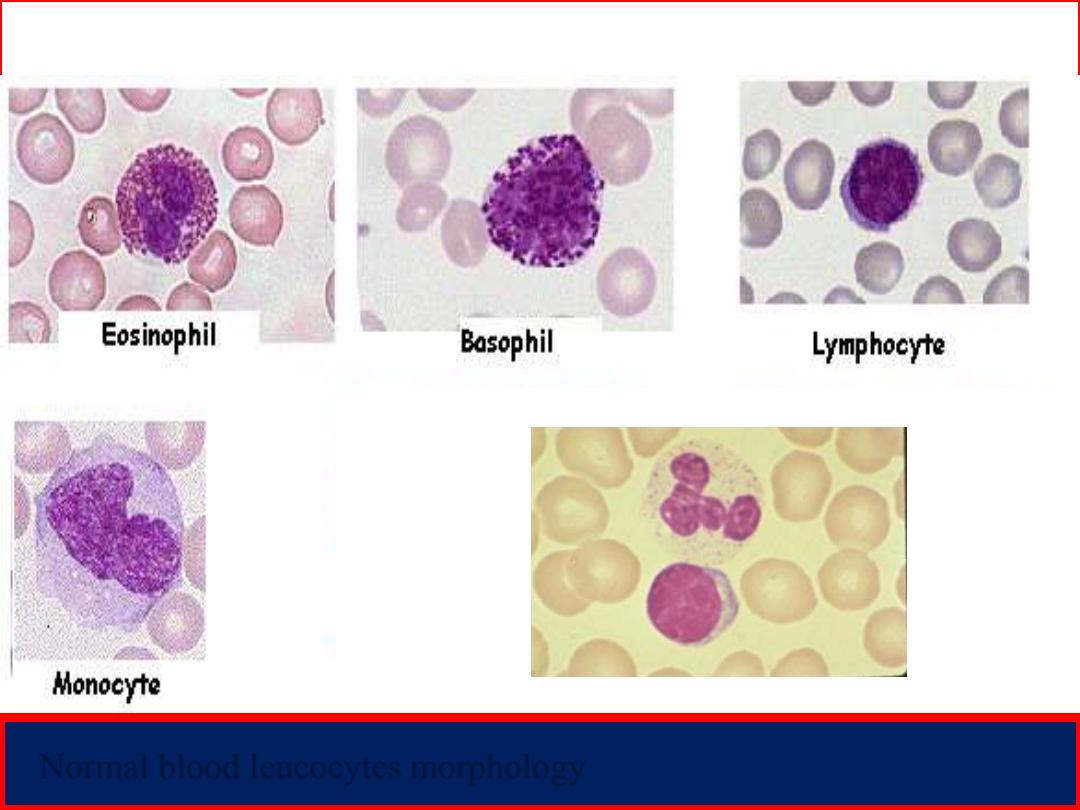
Peripheral blood smear
Normal blood leucocytes morphology
Neutrophil
Lymphocyte
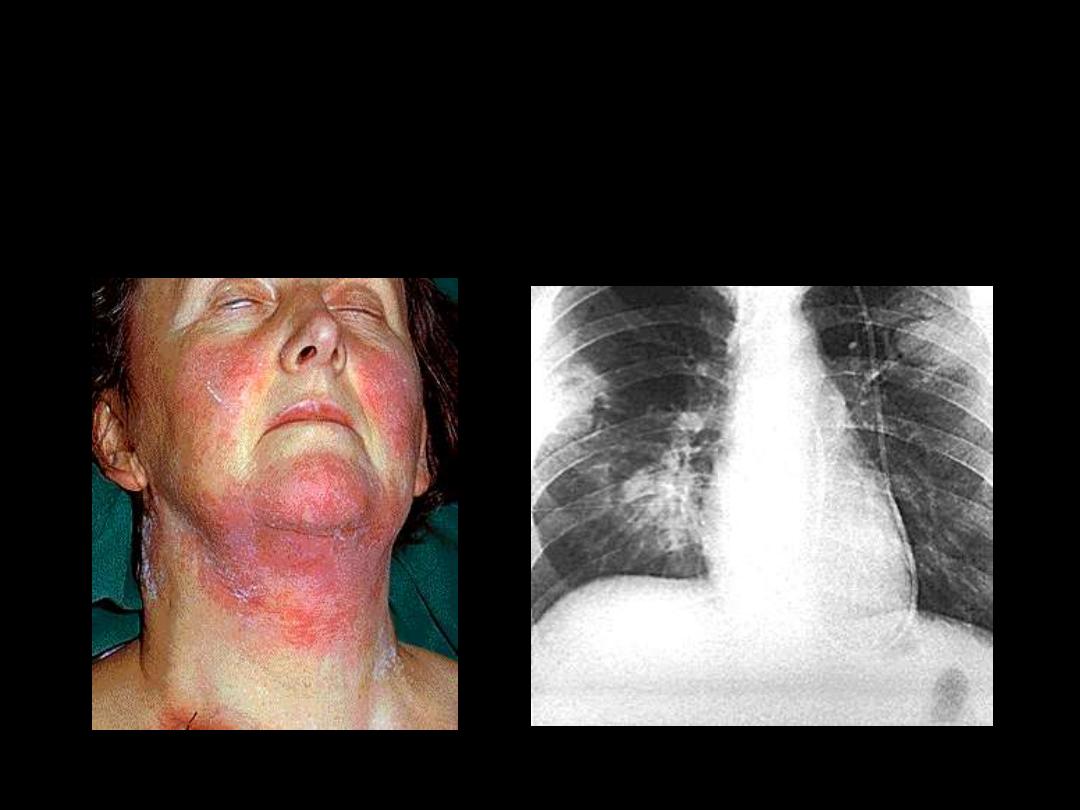
Skin infection Respiratory infection
Neutropenia;
Fever and Infections due to reduced immunity, especially;
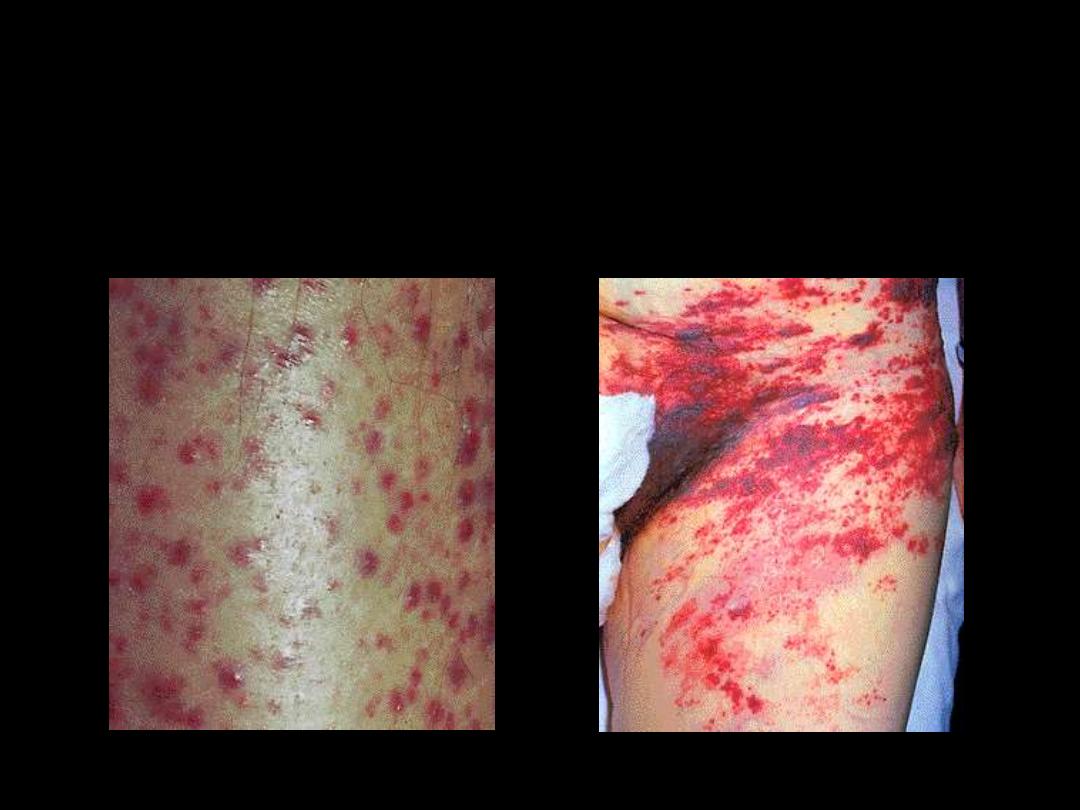
(Ecchymosis)
Thrombocytopenia;
Bleeding manifestations into the skin;
( Petechiae)
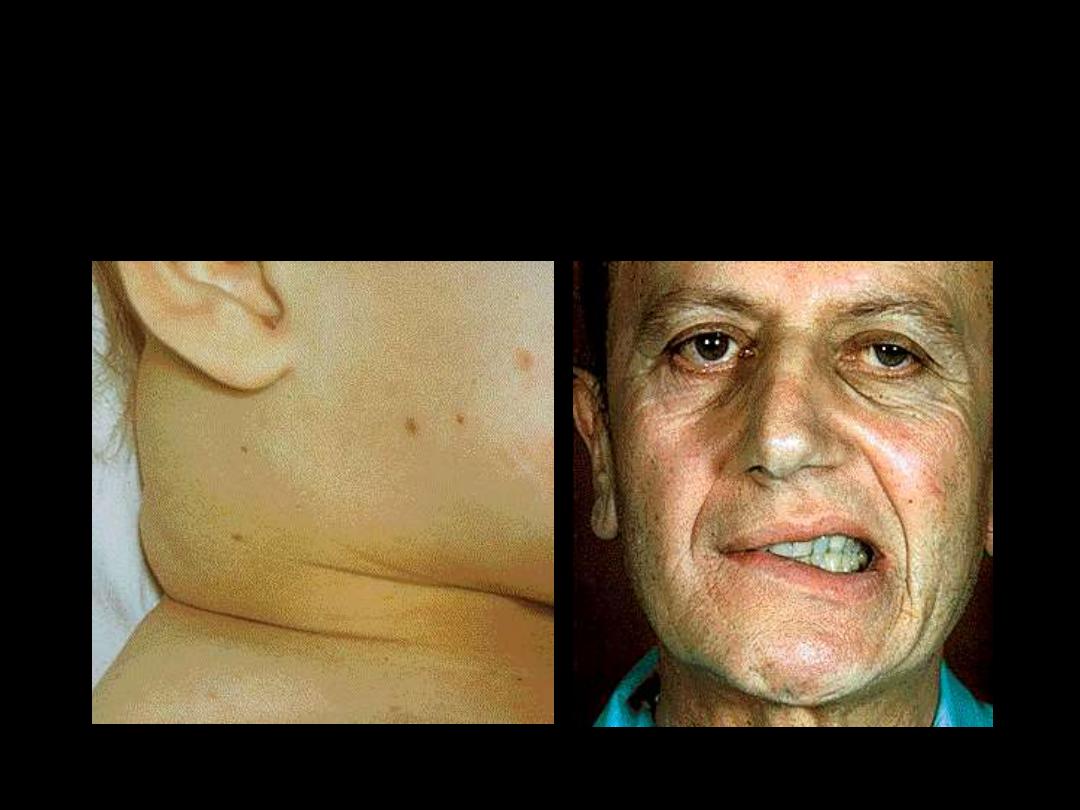
2.
Organ and Tissue Infiltration by the leukemic cells:
Splenomegaly..Hepatomegaly..Bone pain..Arthralgia..
Facial Palsy
Lymphadenopathy
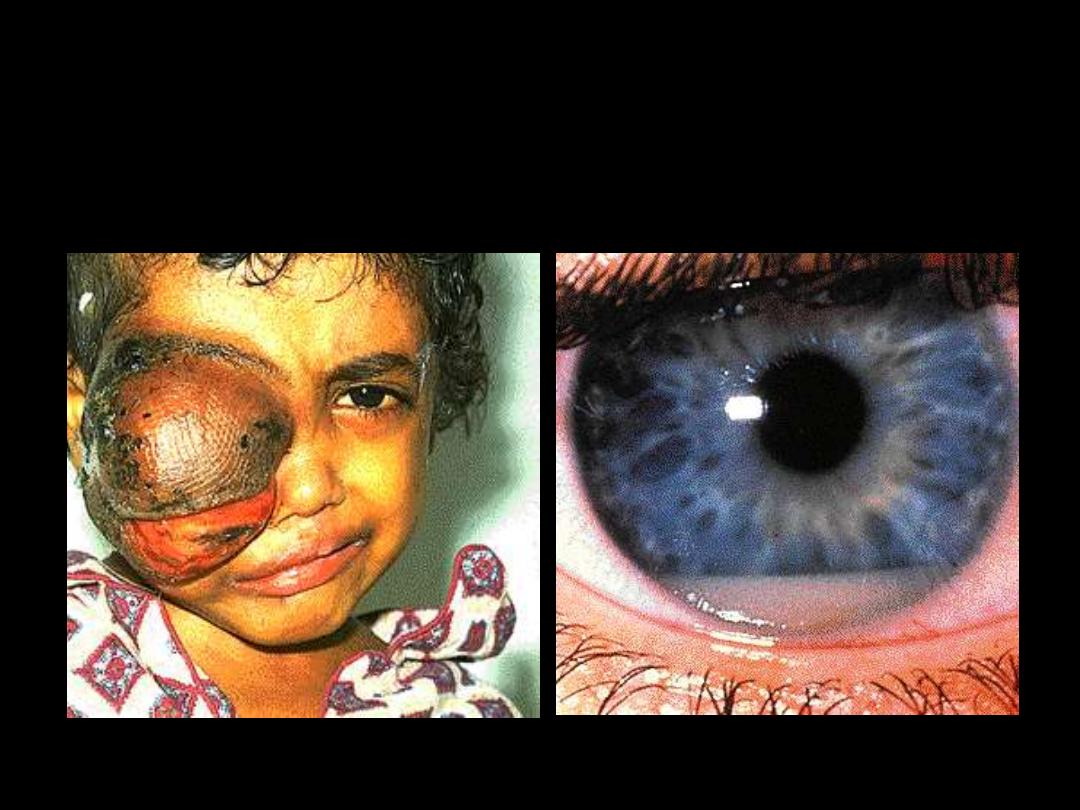
Ant. chamber infiltration
Ocular infiltration
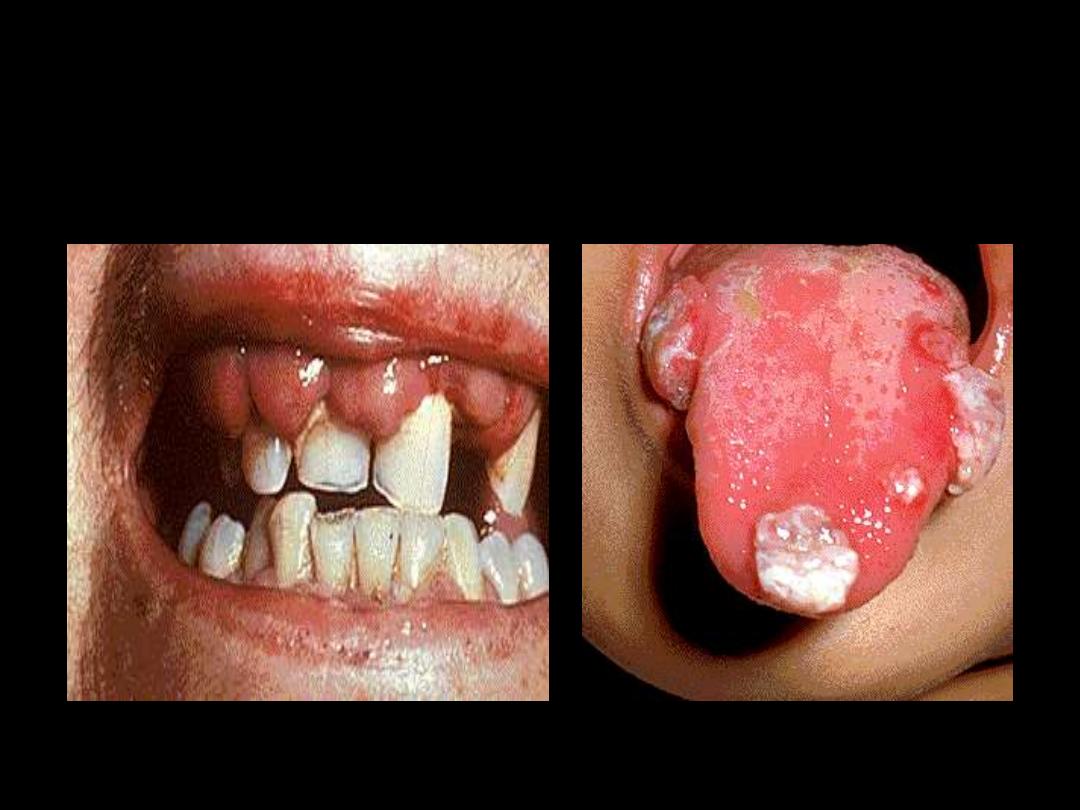
Tongue infiltration
Gum infiltration
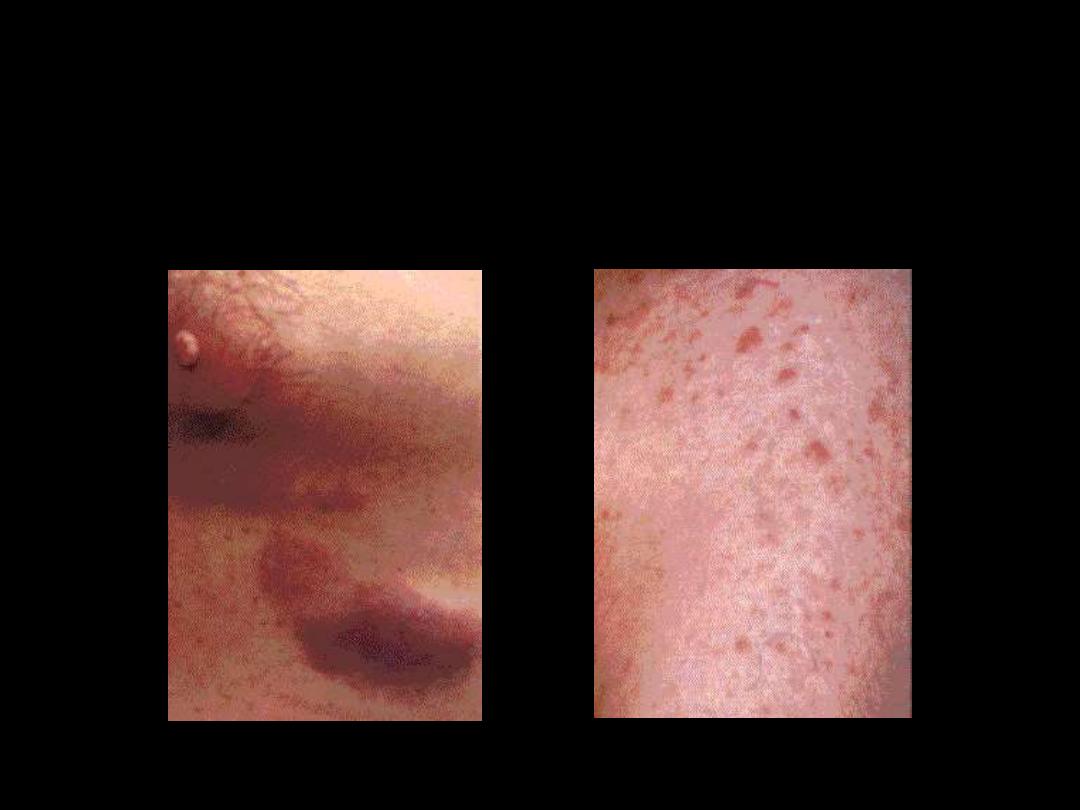
Skin infiltration
Nodular lesion
Raised erythematous lesion
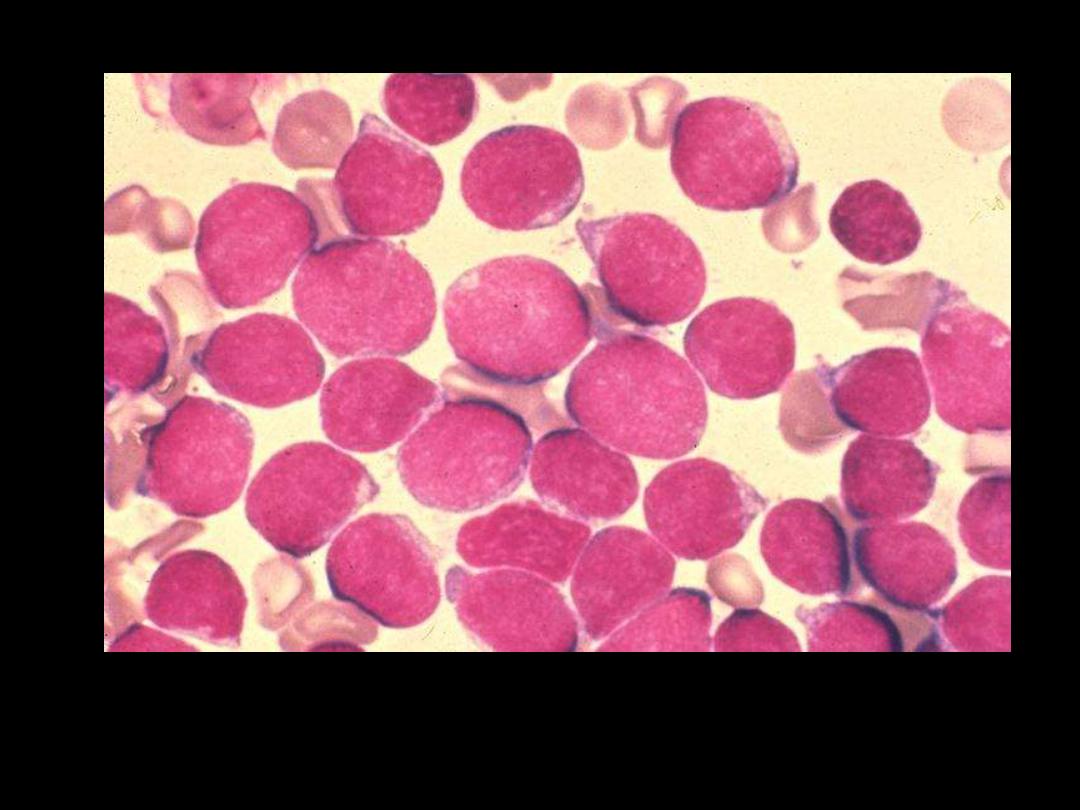
ALL (Lymphoblasts)
The WBC's seen here are lymphocytes, but they are blasts--very
immature cells with larger nuclei that contain nucleoli. Such
lymphocytes are indicative of acute lymphocytic leukemia (ALL
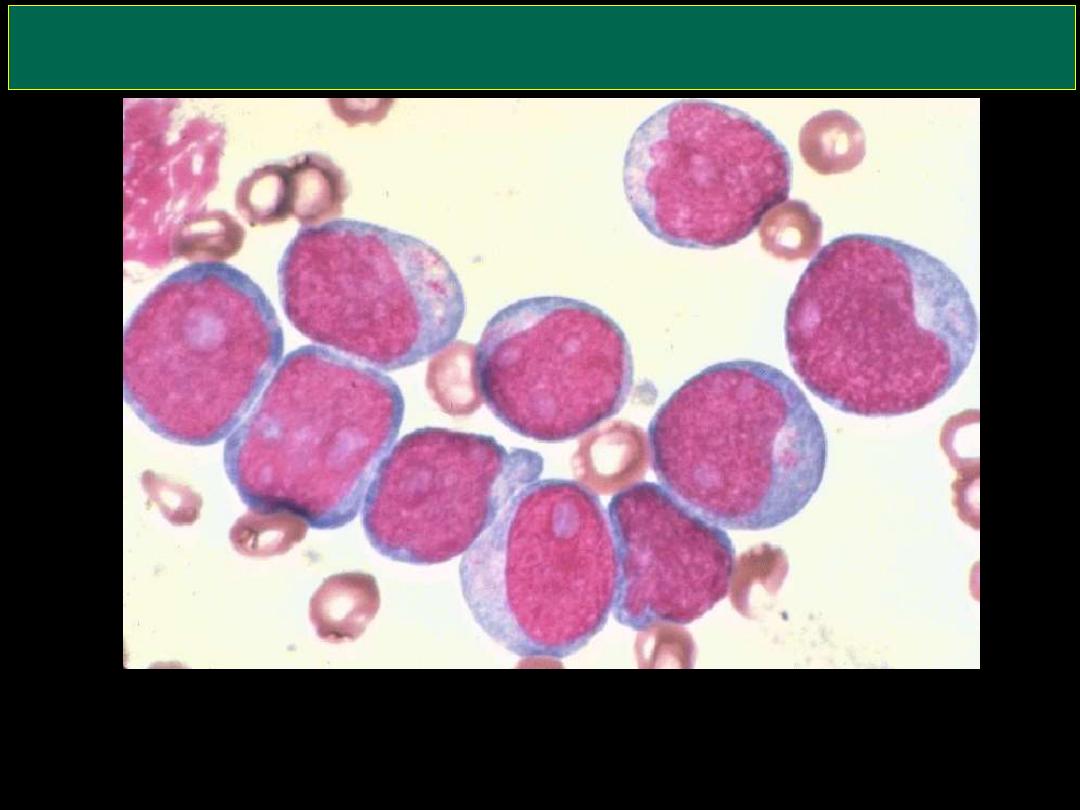
AML (Myeloblasts)
Here are very large, immature myeloblasts with many nucleoli. A distincitve
feature of these blasts is a linear red "Auer rod" composed of crystallized
granules. These findings are typical for acute myelogenous leukemia (AML)
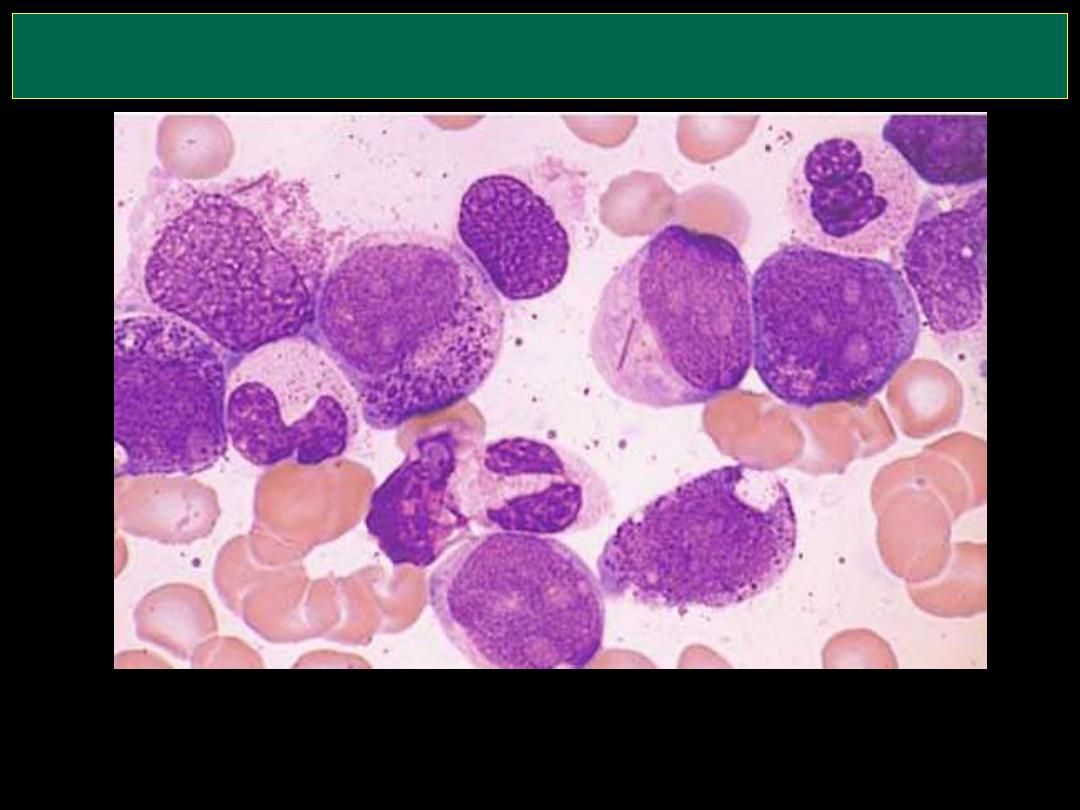
AML (Myeloblast with Auer rod)
Here are very large, immature myeloblasts with many nucleoli. A distincitve
feature of these blasts is a linear red "Auer rod" composed of crystallized
granules. These findings are typical for acute myelogenous leukemia (AML)
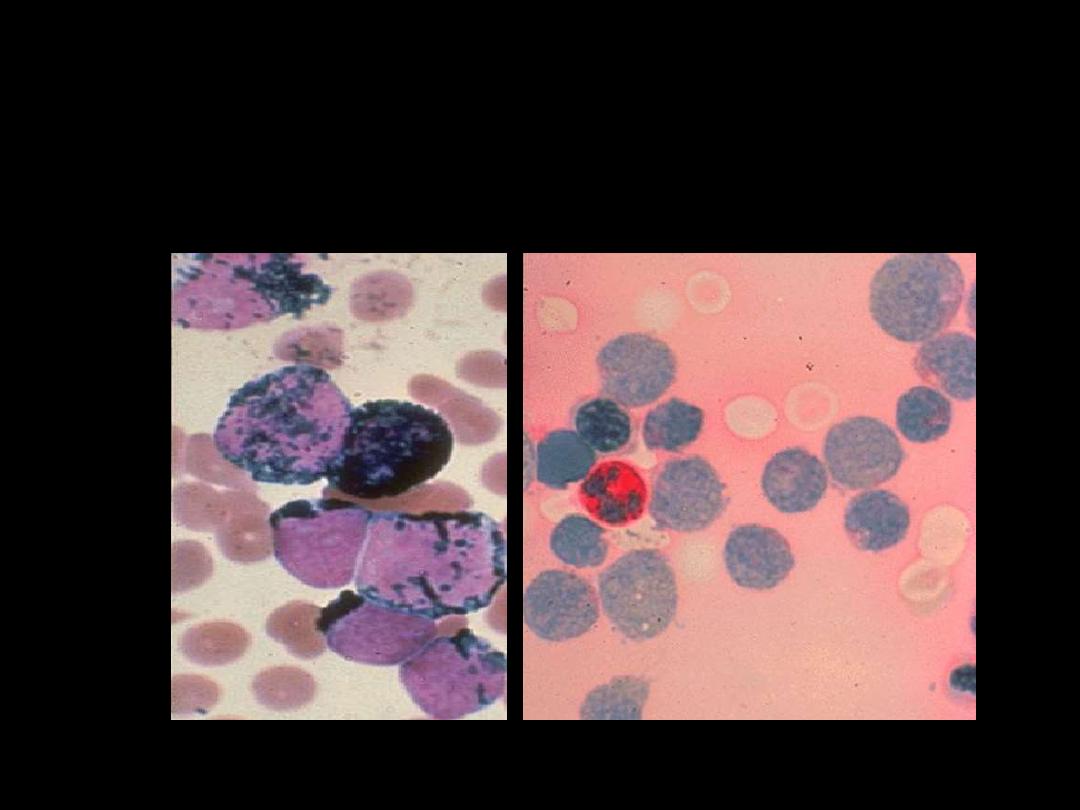
Cytochemistry in AML M1 – M4
SBB positive
PAS negative

LPD
F:\lectures\4th
grade\Pathology\New folder
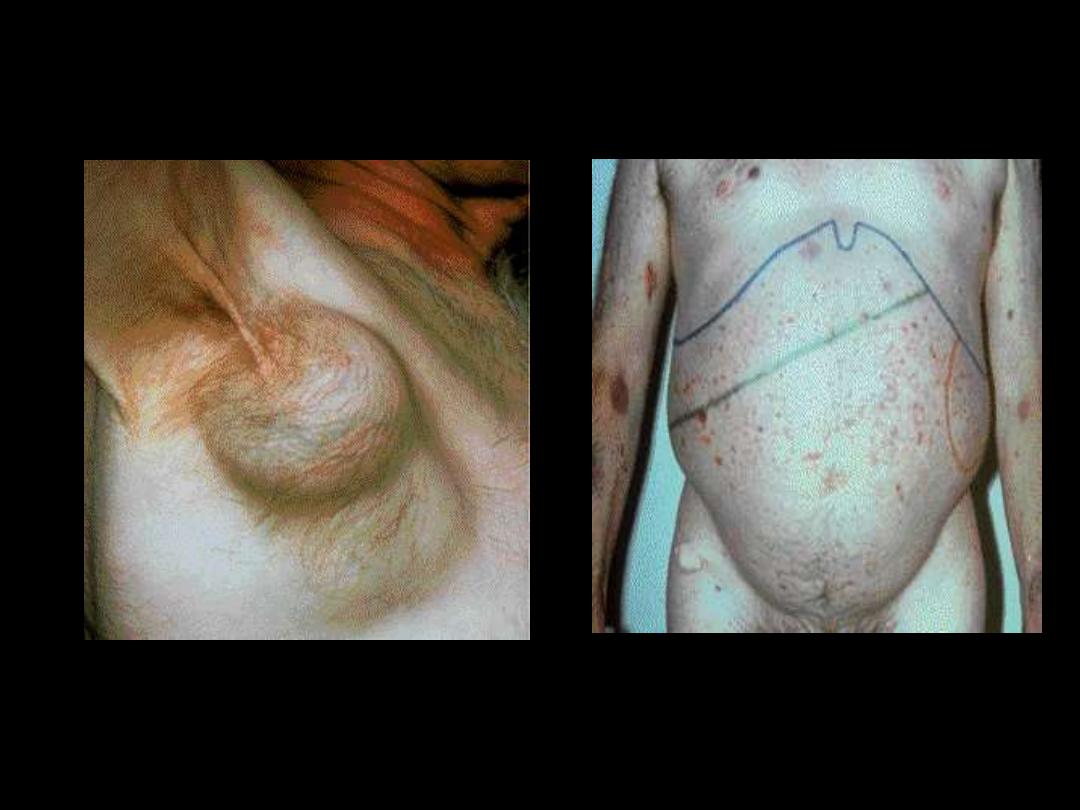
CLL; Axillary LAP
HSM with purpura &
ecchymosiis
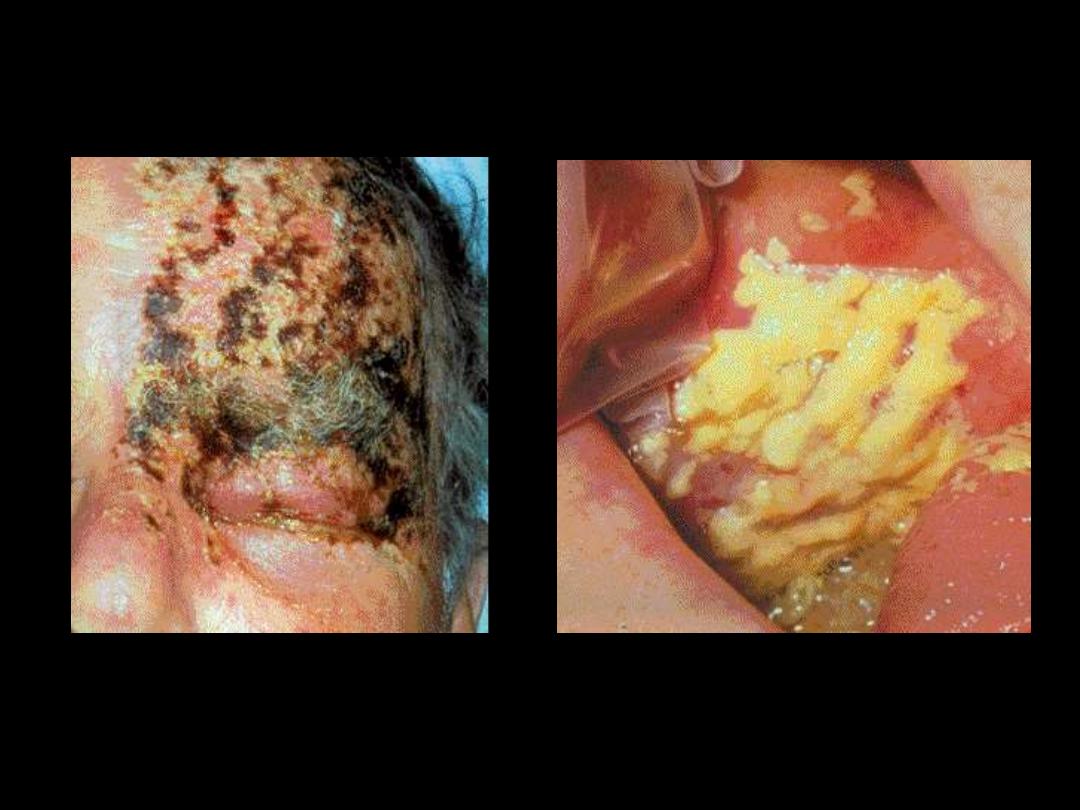
CLL; Herpes zoster
Buccal Cavity: Candida
albicans
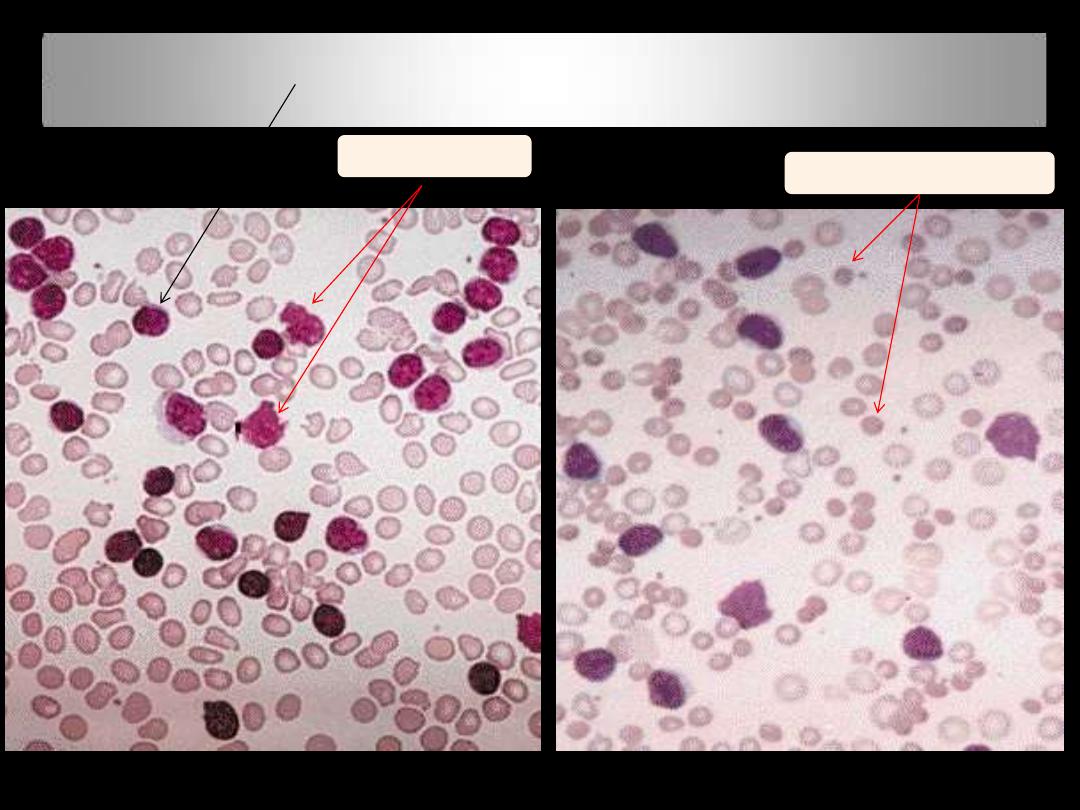
PBF in CLL: Small mature appearing lymphocytes with
compact chromatin and scanty cytoplasm
Smudge
cells
Spherocytes in
AIHA
CLL; BF
CLL/PL
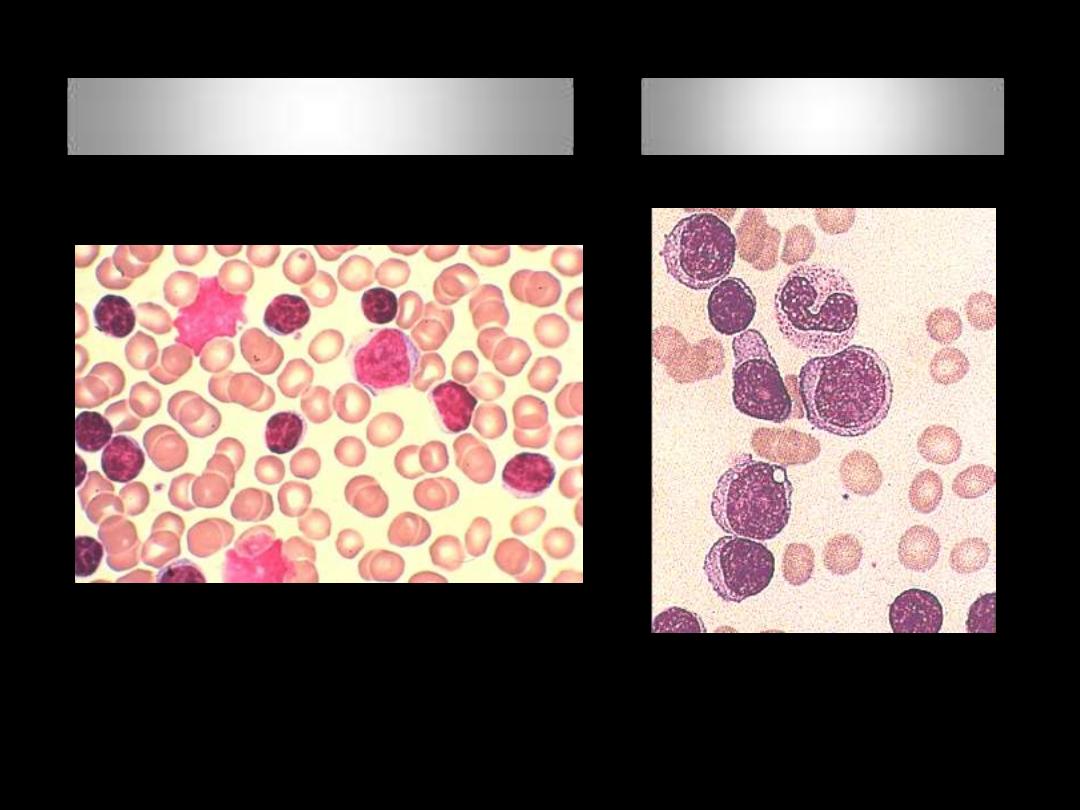
Richter Transformation
(BF)
Richter Transformation
(LN)
Prolymphocytic leukemia PLL; BF
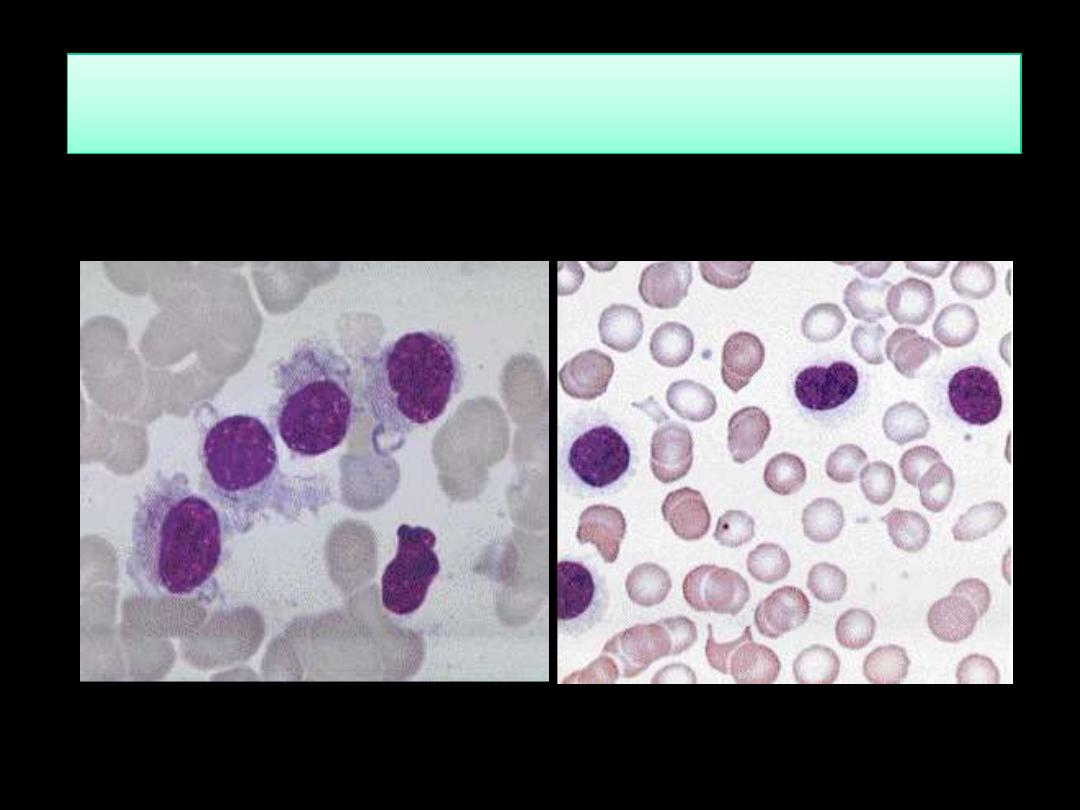
Hairy Cell leukemia HCL; BF

MPN
F:\lectures\4th
grade\Pathology\New folder
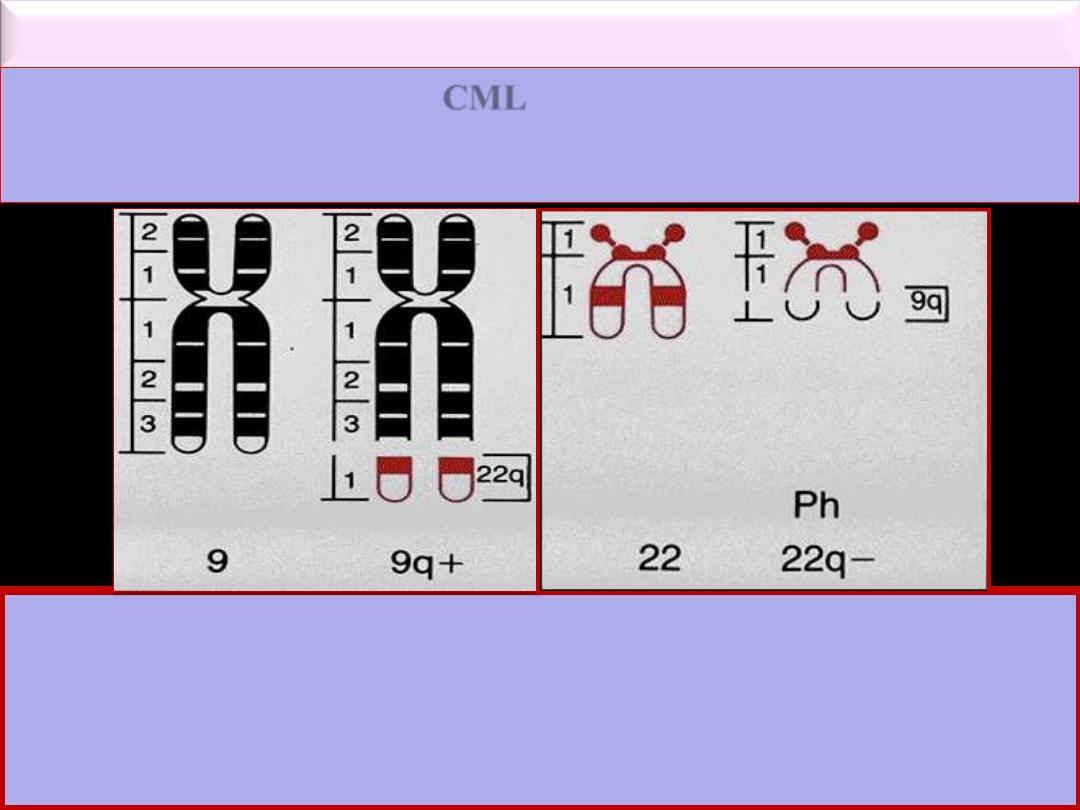
Karyotype and Molecular Features
The vast majority of
CML
show the Philadelphia (Ph
+
)
chromosome* in (90-95%) and M-BCR-ABL p210 in (99% of
patients), these two discoveries in 1960 & 1986 respectively are
important landmarks.
*
Ph chromosome
is a minute chromosome 22 from which the long arms are
deleted (22q-). It is part of reciprocal translocation between chromosome 9
& 22 t(9; 22)(q34; q11) in which part of 22 is clearly visible on 9 but the
part of 9 on 22 is too small to be distinguished cytogenetically. This
translocation is detected by PCR or FISH techniques
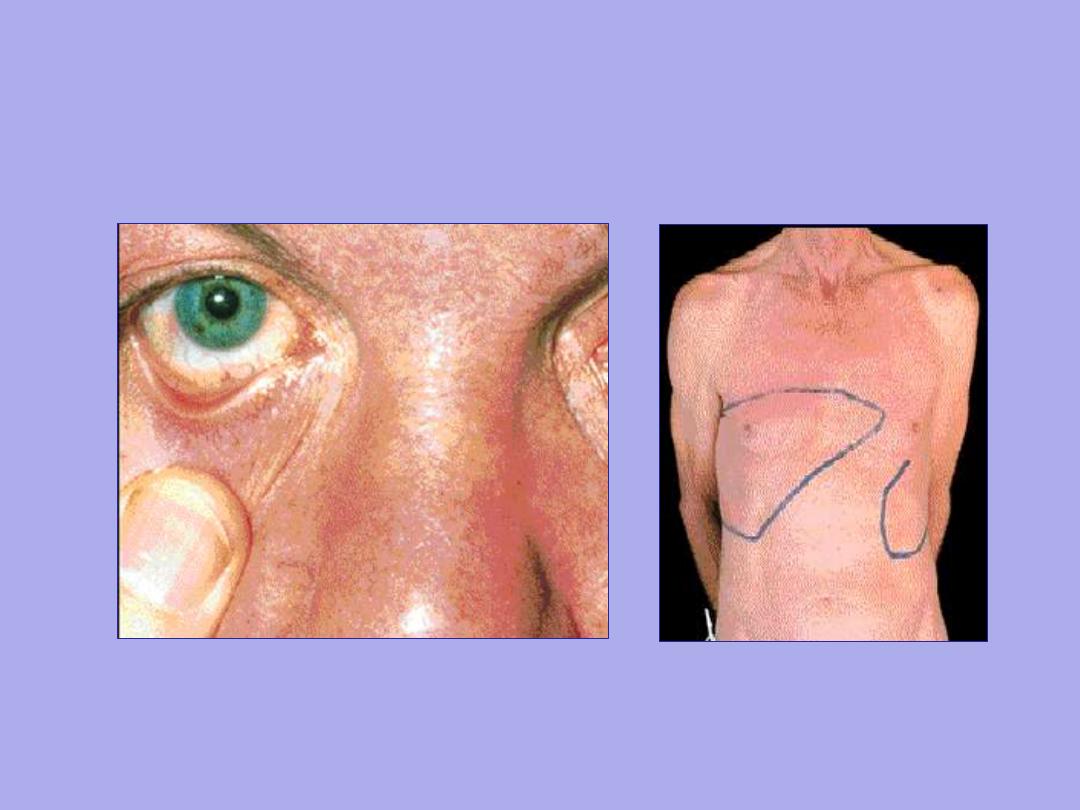
Conjunctival suffusion Hepatosplenomegaly
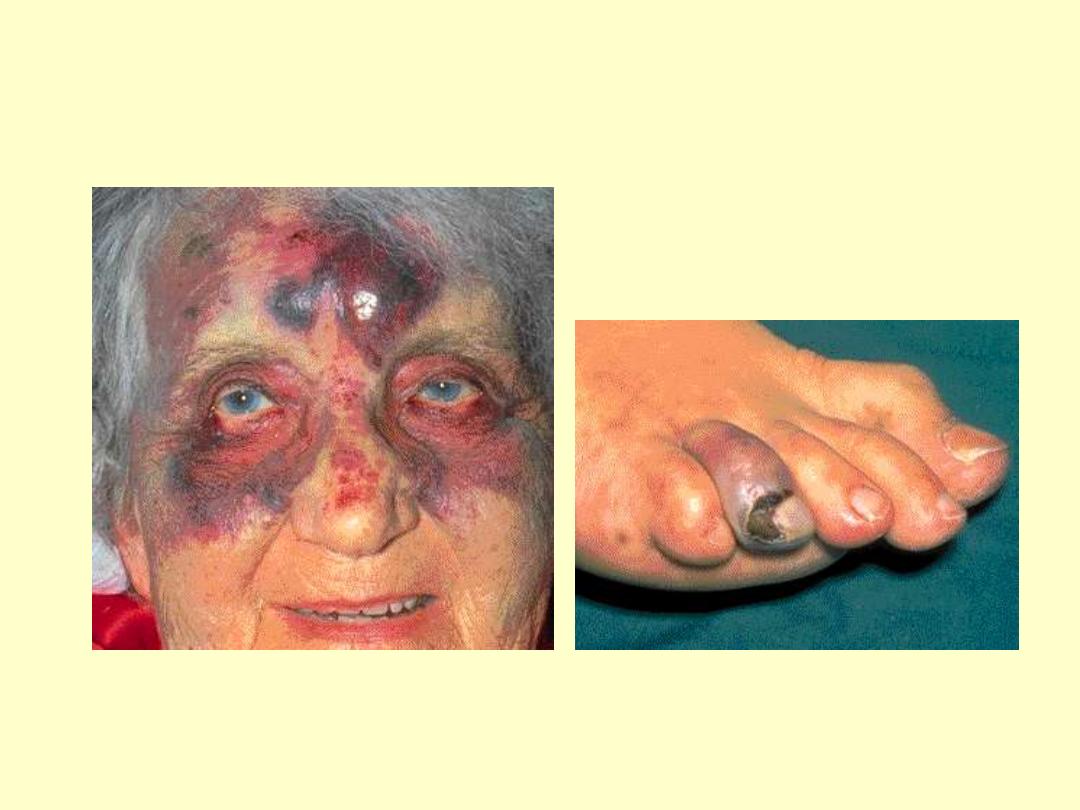
Essential Thrombocythemia
Hemorrhage after minor trauma
due to platelet dysfunction
Gangrene of the toe
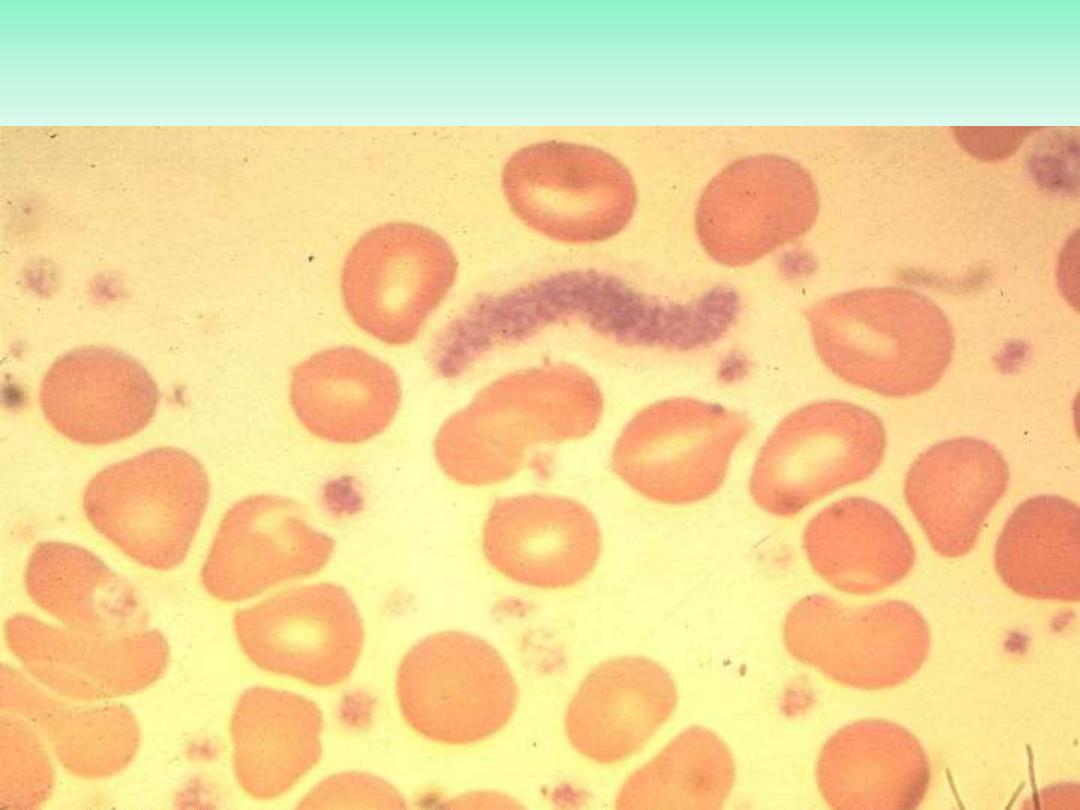
Essential thrombocythemia blood film
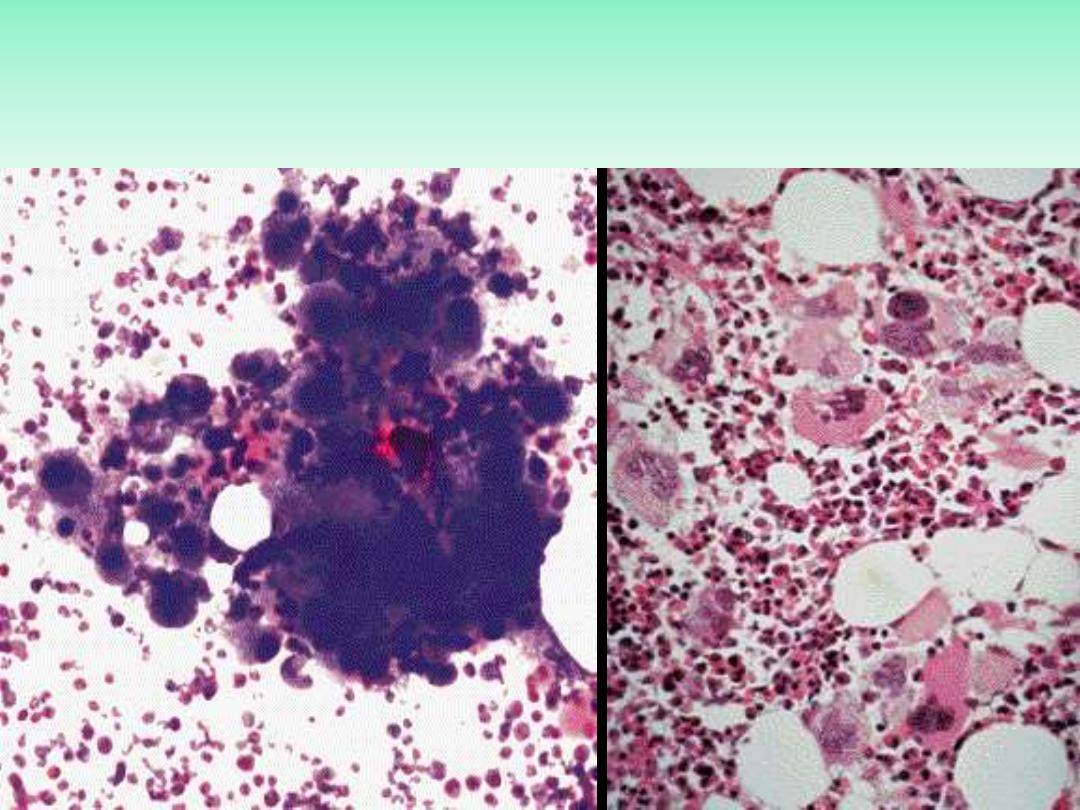
Essential thrombocythemia
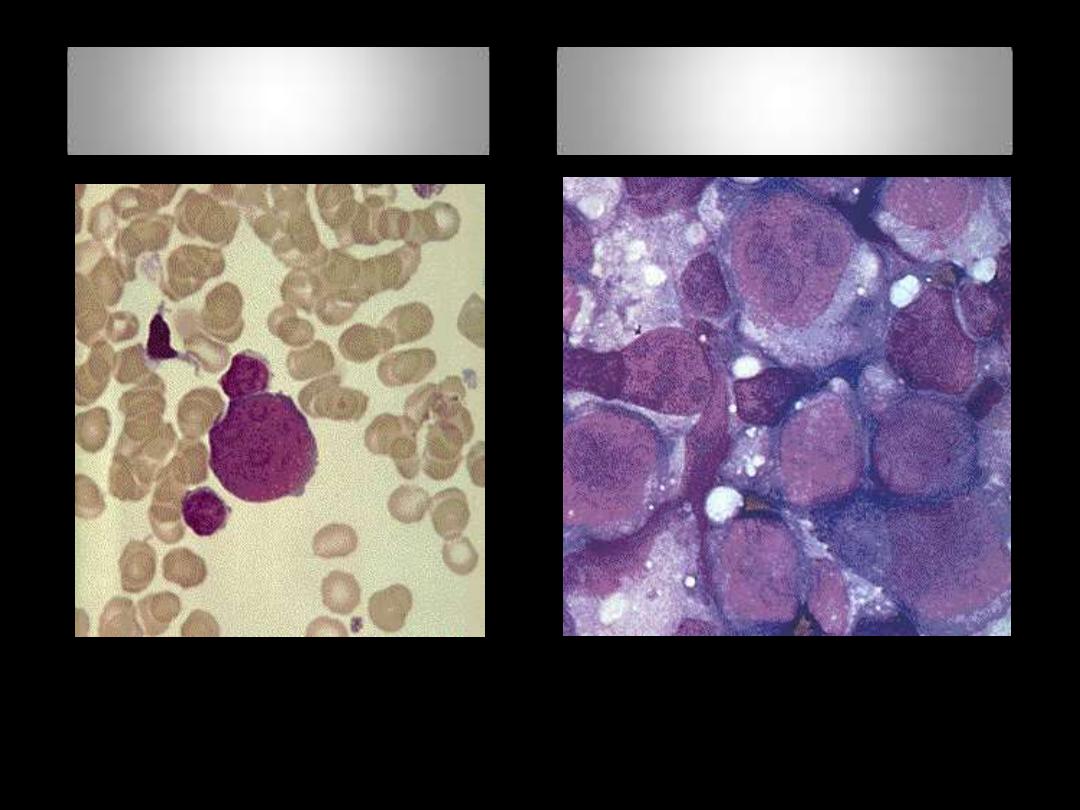
BMA (Left), BMB (Right)
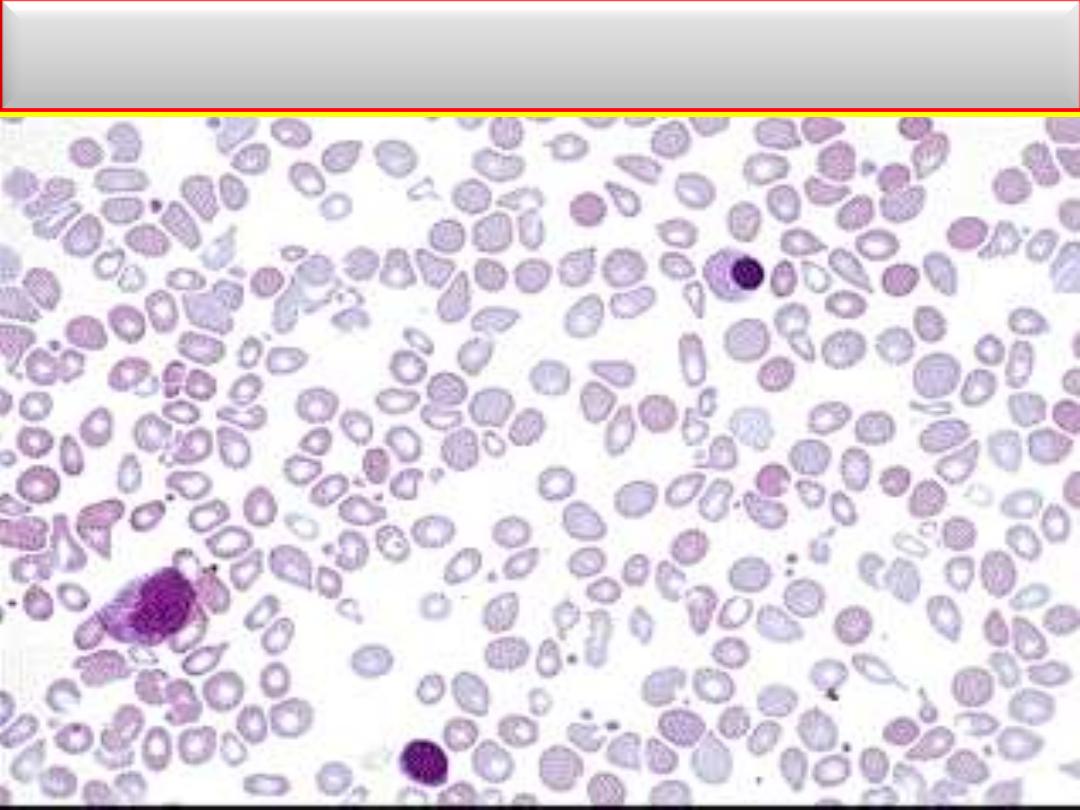
Laboratory Findings: MF
BF: Tear drop cells with leukoerythroblastic blood picture
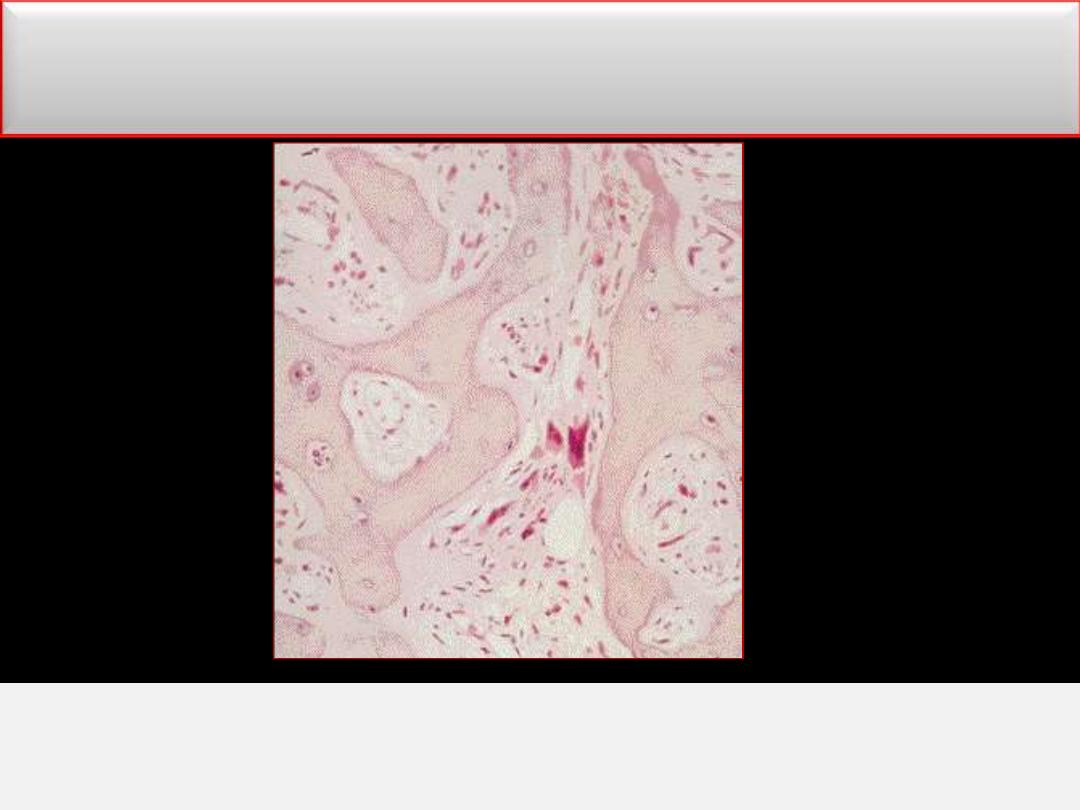
Myelofibrosis
BM biopsy in advanced disease
OsteoMF, replacement by fibrous connective tissue with thick bone
trabeculae
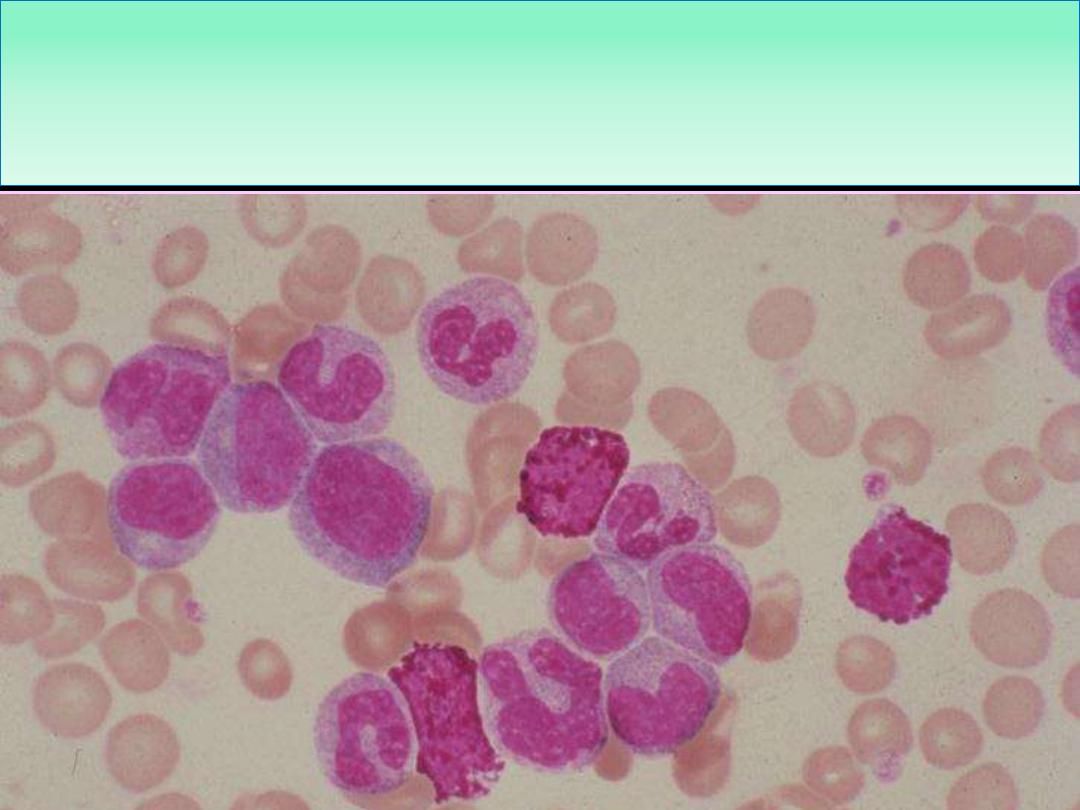
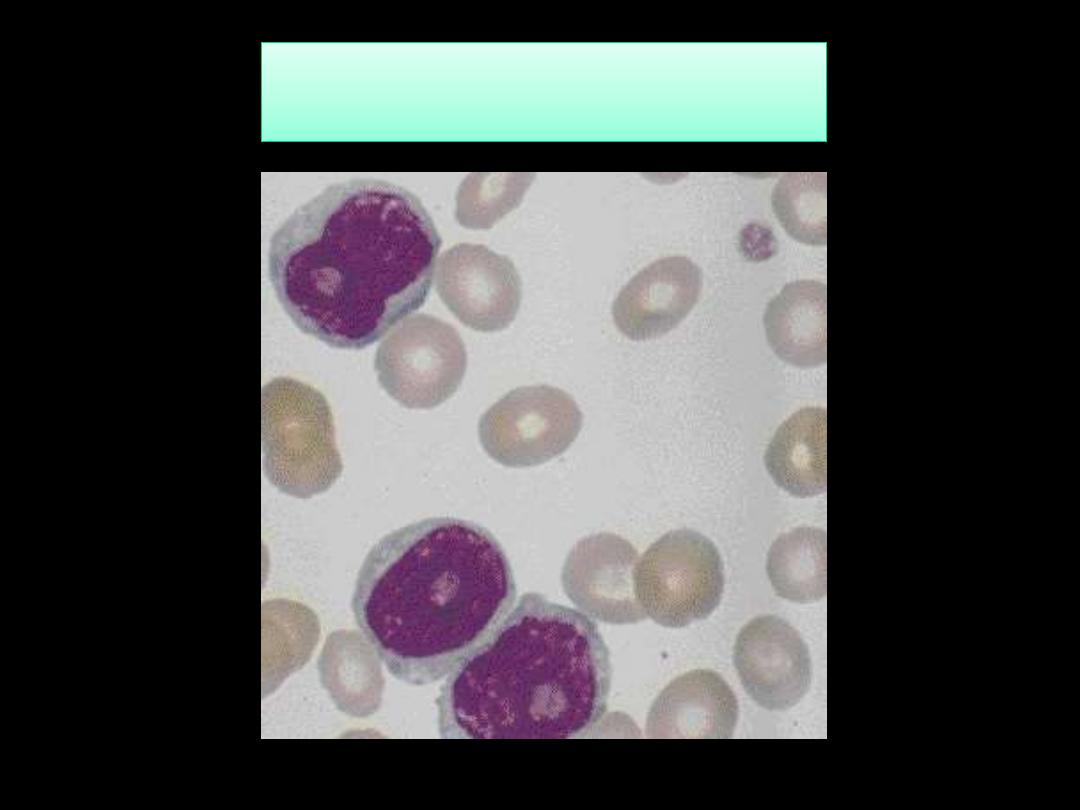
Blood Film in CML - CP showing broad spectrum of granulocytic cells
in various stages of maturation (Promyelocytes, myelocytes,
metamyelocytes, neutrophils basophils and a monocyte)
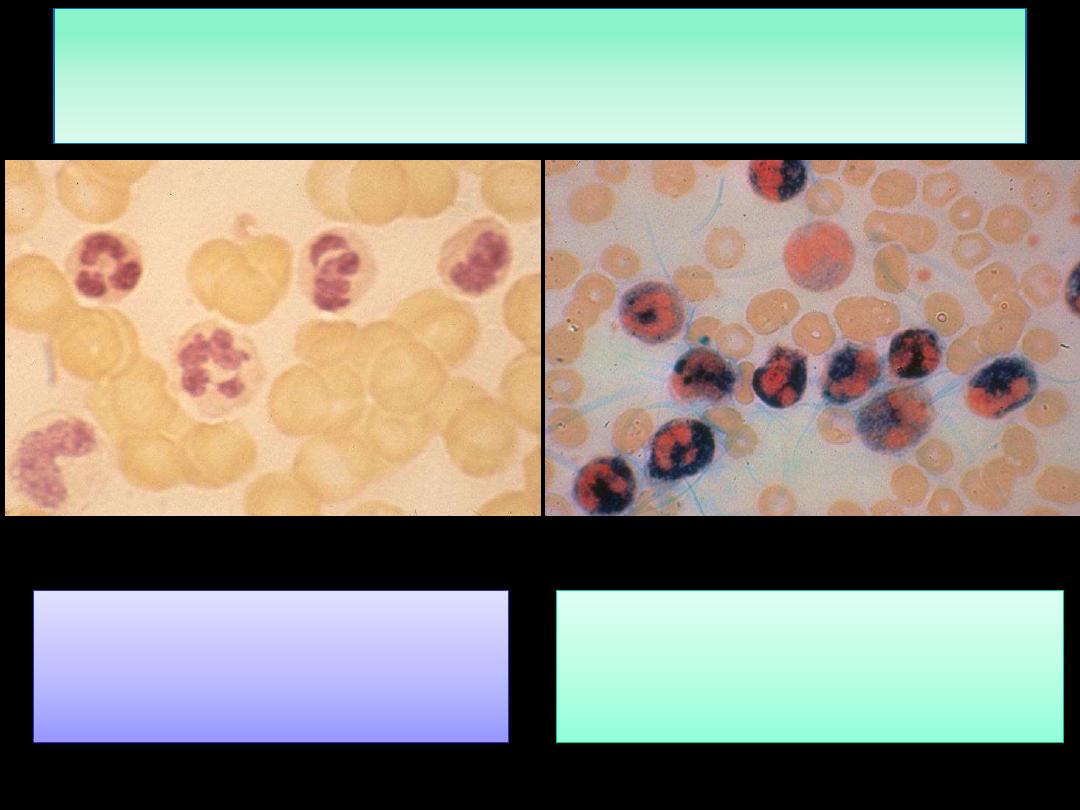
Laboratory Findings:
Absent NAP score (it is low
or absent in about 95% of
CML cases)
Increased NAP score, as is seen
by the dark staining reaction
product in the neutrophils

PCN
F:\lectures\4th
grade\Pathology\New folder
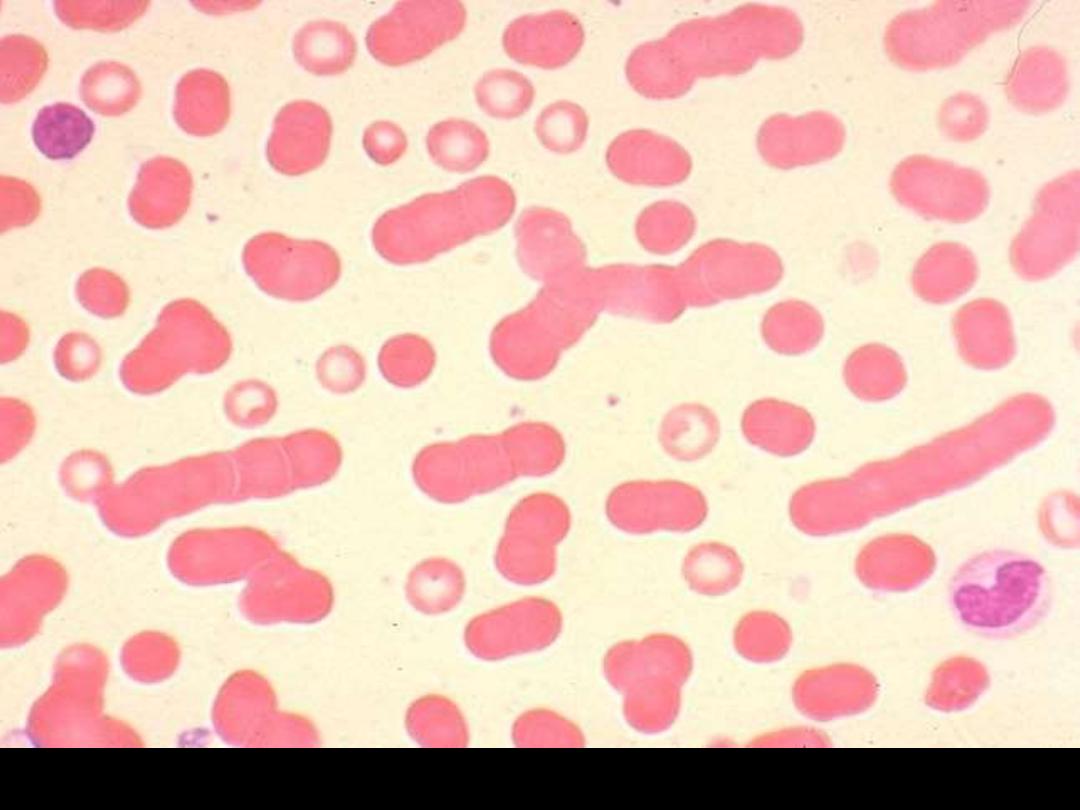
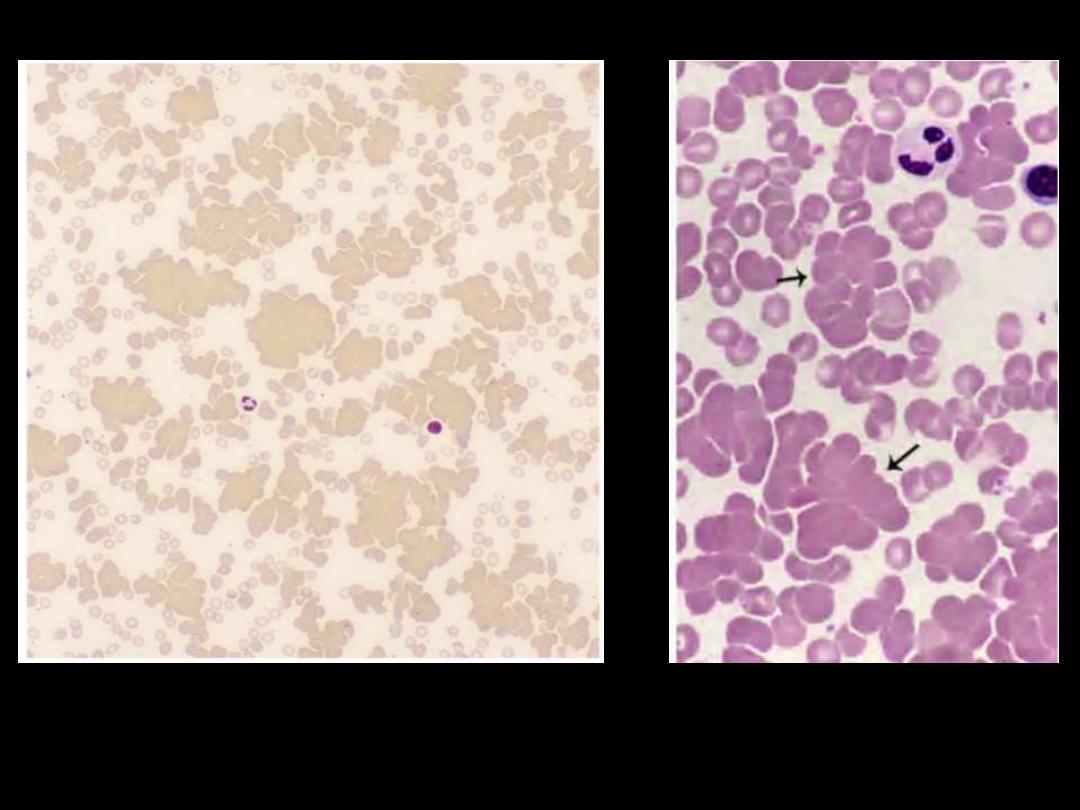
Blood film showing marked rouleaux formation
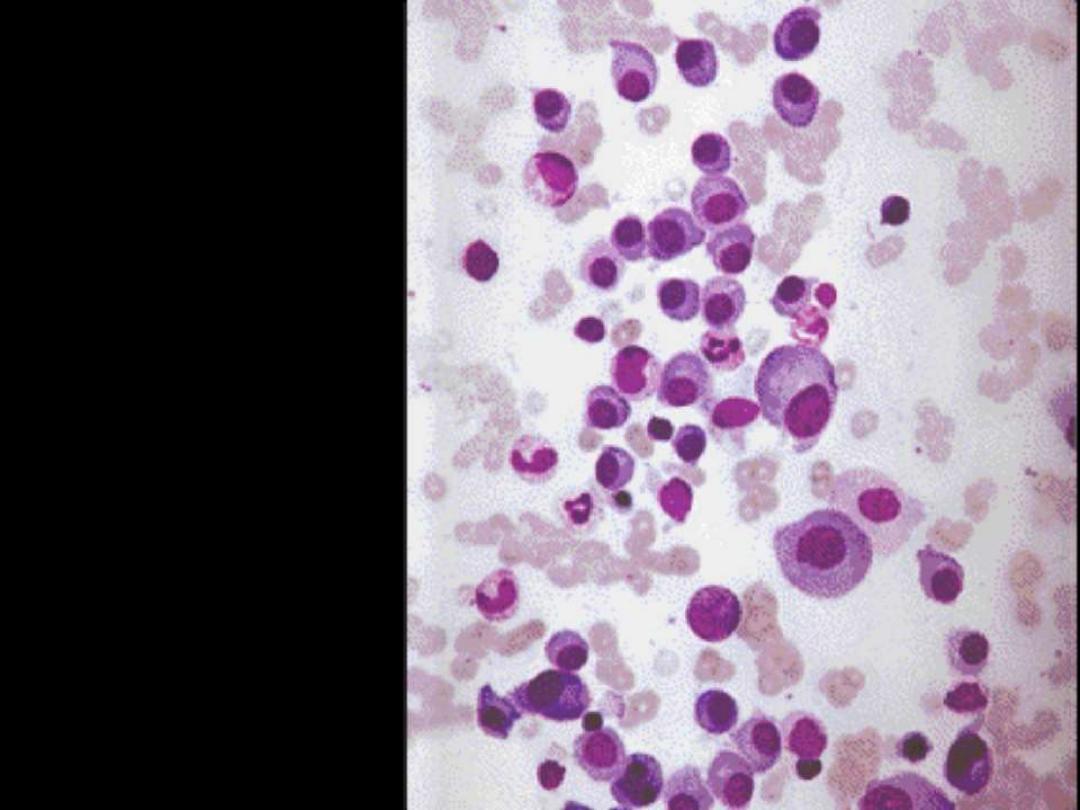
16
Bone marrow showing
extensive infiltration by
plasma cells
Describe the changes
in the BM smear?
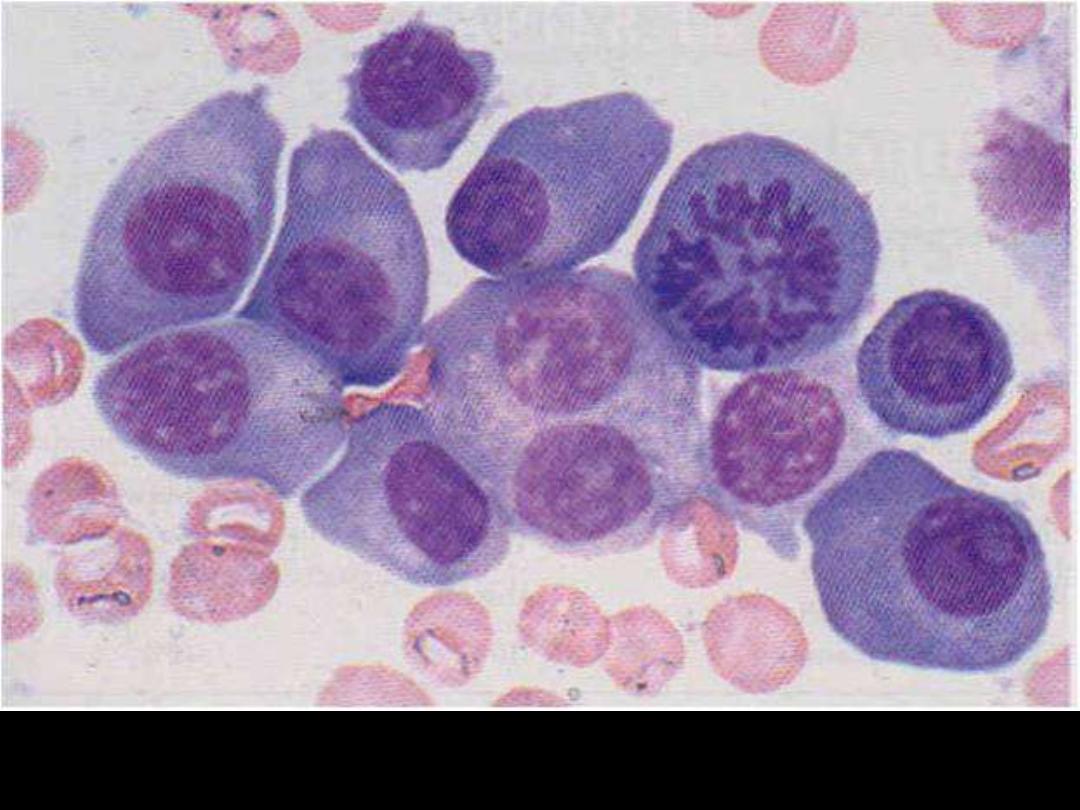
Increased number of plasma cells in the BM
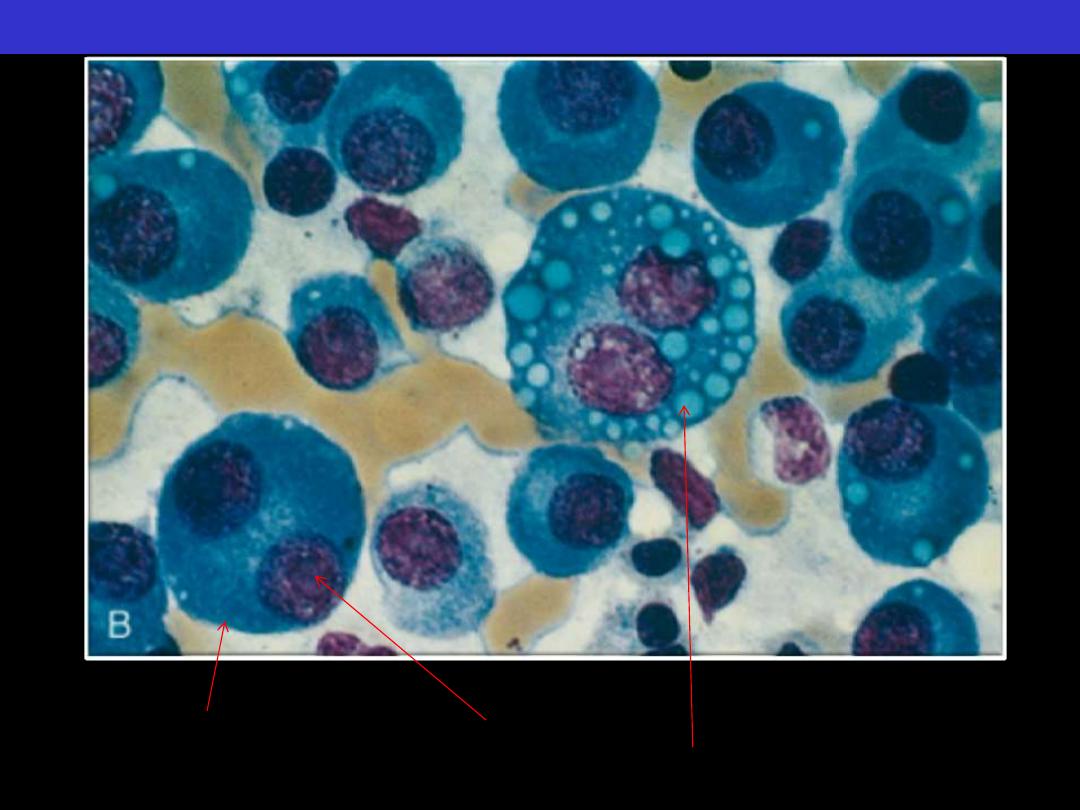
Normal marrow cells are largely replaced by plasma cells, including
atypical binucleated forms, prominent nucleoli, and cytoplasmic droplets
containing immunoglobulin.
Multiple myeloma BM aspirate
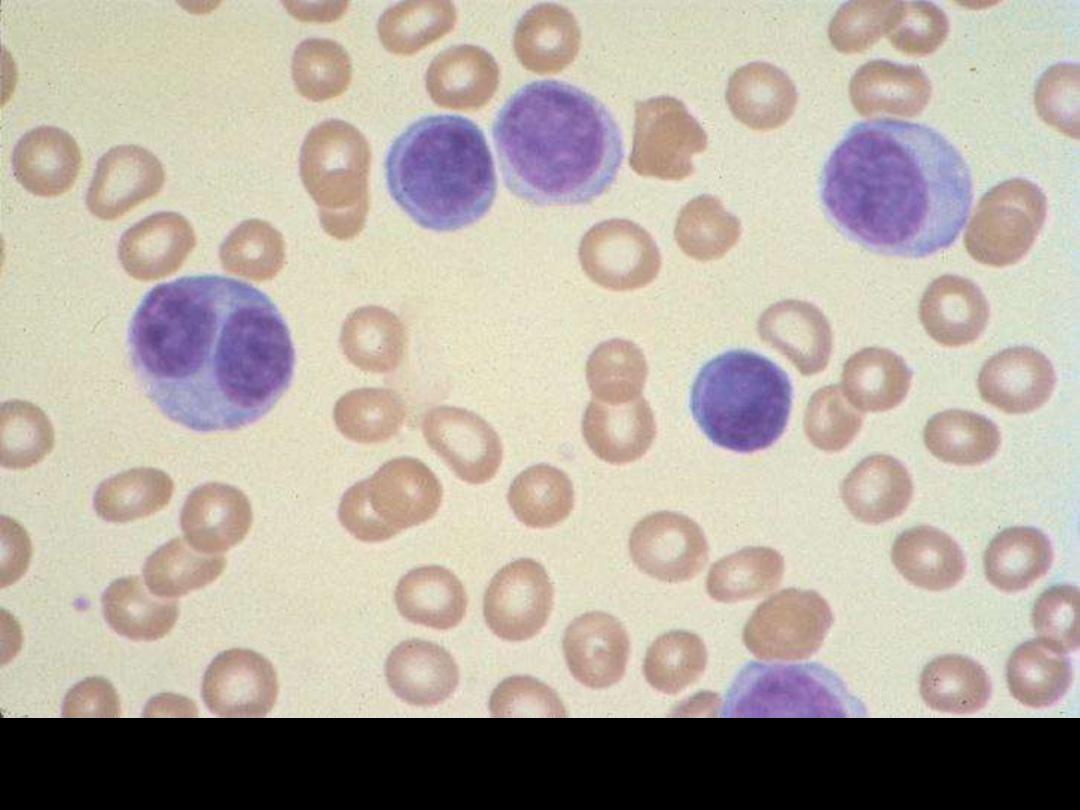
Plasma cell leukemia
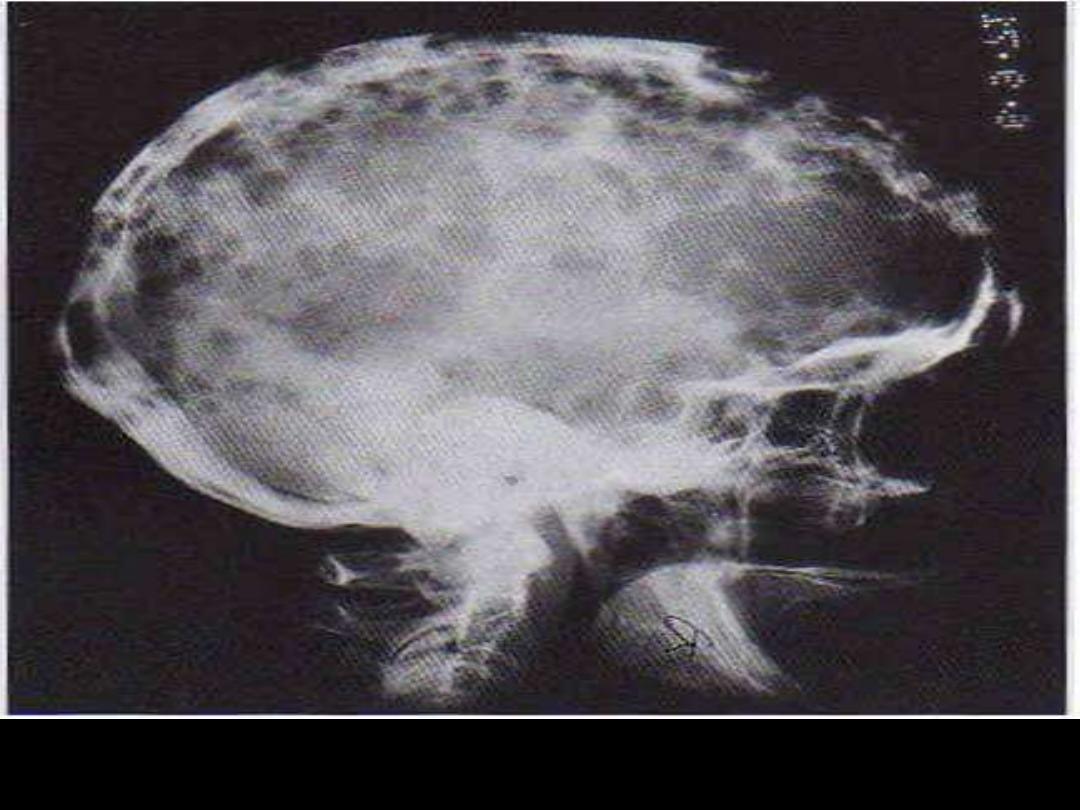
Skull X-ray: Osteolytic punched-out lesions are most obvious in the
calvarium.