
Neuro
Dr. Nibal
“ Acute Flaccid Paralysis ”
Total Lec: 45


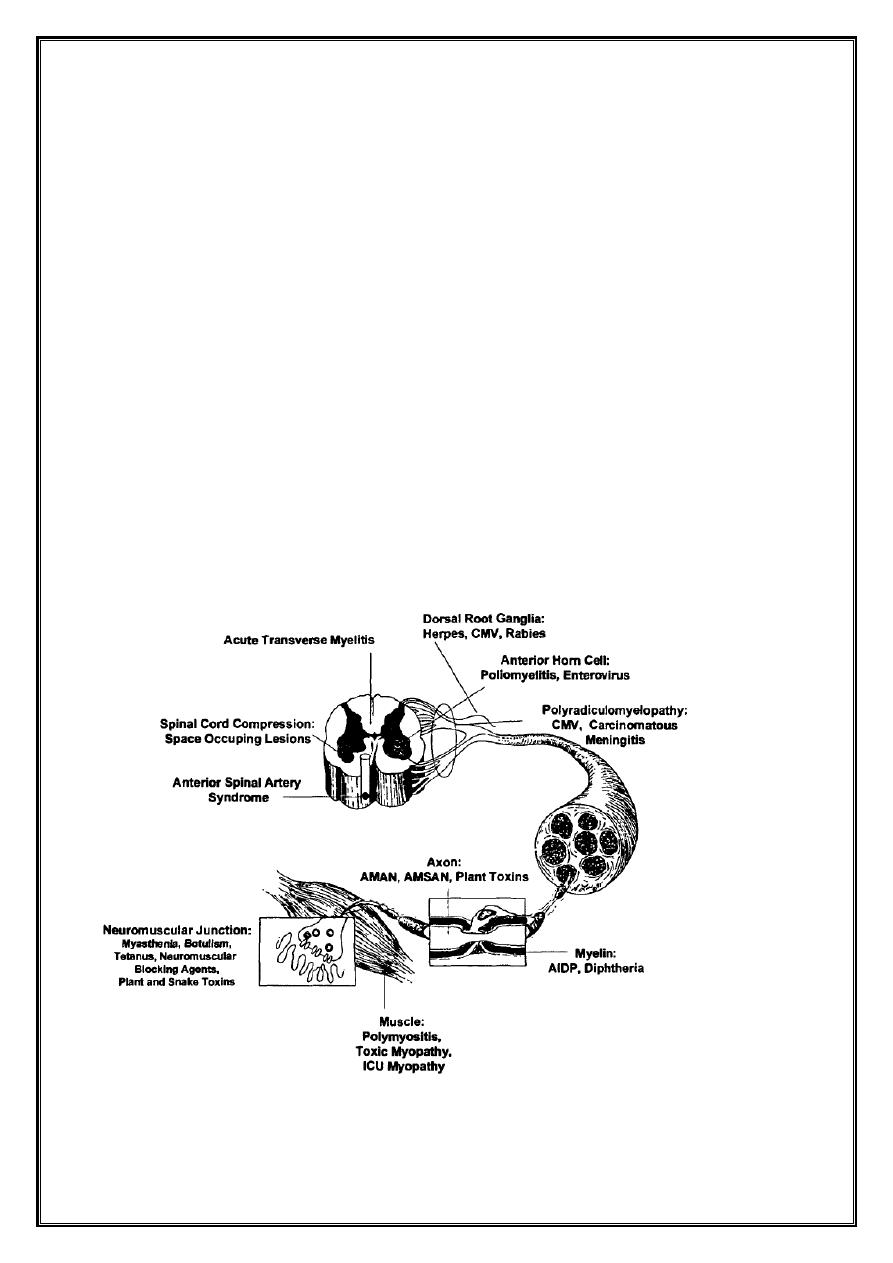
Acute flaccid paralysis
Dr Nebal Waill
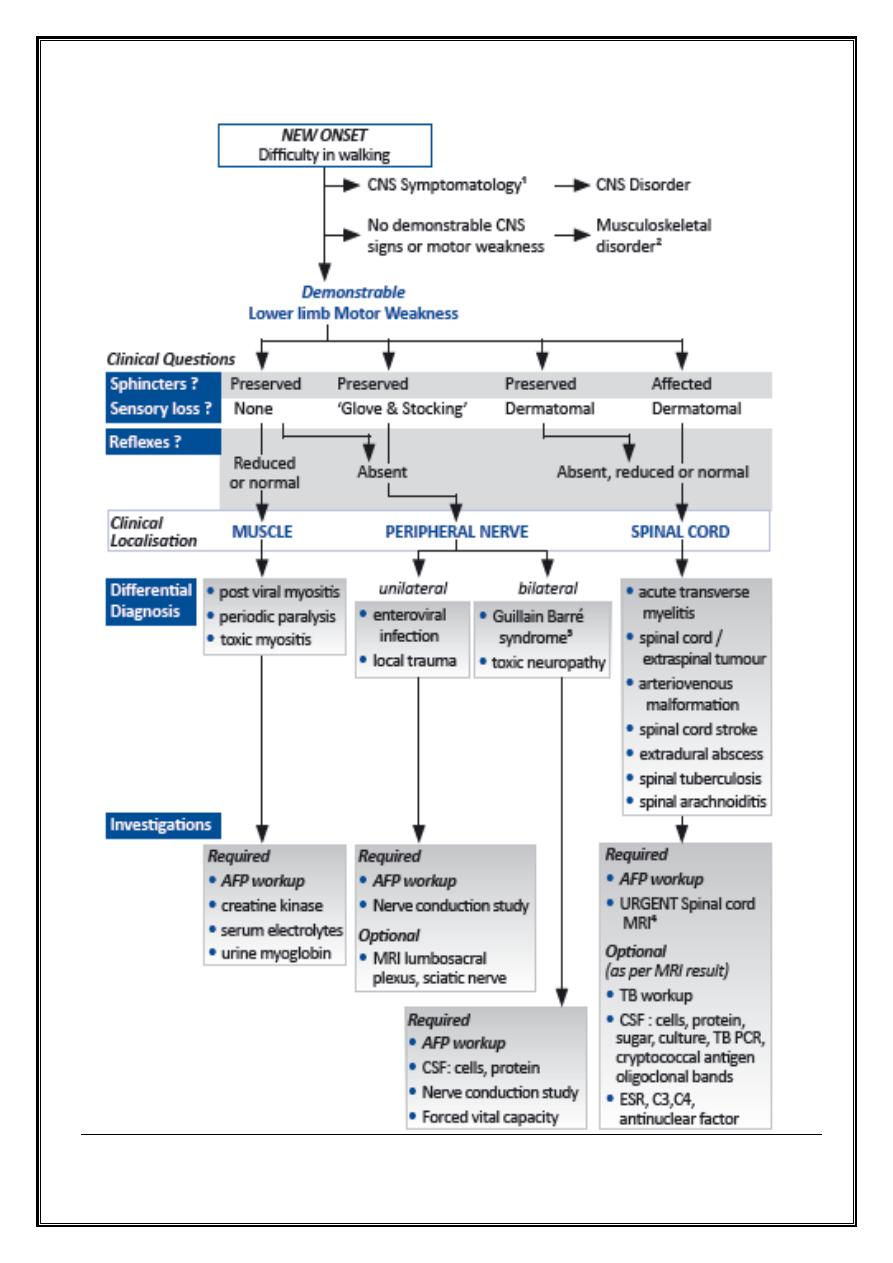
Definition
•
Acute Flaccid Paralysis (AFP) occurs when there is rapid evolution of motor
weakness (< than 4 days), with a loss of tone in the paralysed limb. This
excludes weakness due to trauma and spastic paralysis.
•
AFP is a medical emergency as unnecessary delays can result in death and
disability

•
The list of underlying causes of AFP is broad, and there is substantial variation
by age, ethnicity, and geographic area.
•
In the absence of wild virus-induced poliomyelitis, the acute demyelinating
form of Guillain-Barre syndrome (AIDP) accounts for at least 50 percent of AFP
cases globally followed in frequency by paralytic non-polio enterovirus
infection, the motor axonal form of Guillain-Barre syndrome (AMAN),
traumatic neuritis, and acute transverse myelitis.
Background
• 1916- Guillian, Barre and Strohl described 2 French soldiers with motor weakness,
areflexia, and “albuniocytological dissociation” in the cerebrospinal fluid. They
recognized the peripheral nature of the illness.
Epidemiology
• Annual incidence of GBS = 1-3/ 100000 persons annually.
• Rare in infants.
• Male & female have similar risk
• Any age but most frequent at 4-9 years
GBS subtypes
1.
Sporadic AIDP (Acute Inflammatory Demyelinating
Polyradiculoneuropathy)
2.
AMSAN (Acute Motor and Sensory Axonal Neuropathy)
3.
AMAN (Acute Motor Axonal Neuropathy )
4.
MFS ( Miller Fisher Syndrome )
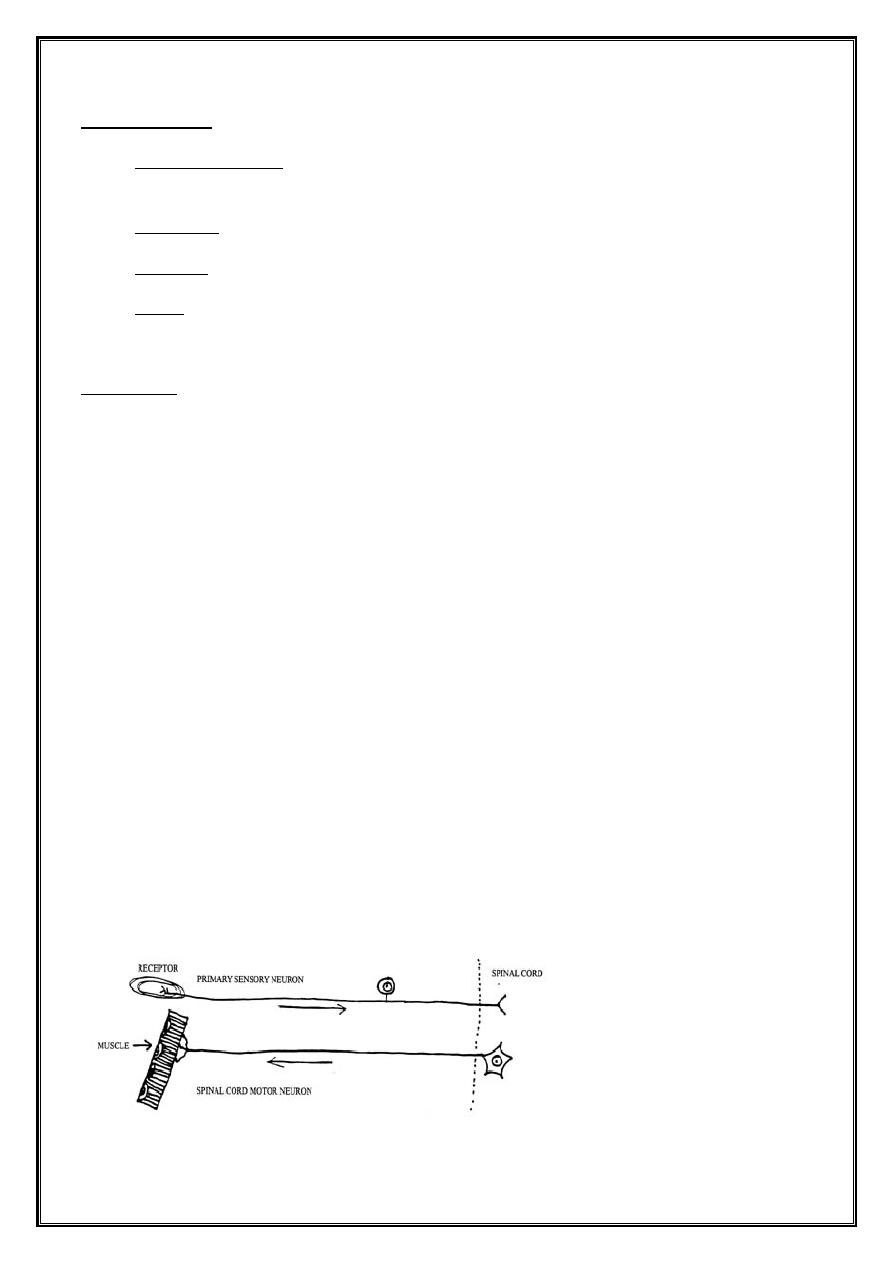
Pathology
•
Both motor and sensory fibers are affected
•
AIDP → segmental demyelination
↘ axonal degeneration ( less extensive )
•
Segmental demyelination occurs at all levels of peripheral nervous system
↘ Anterior + posterior roots
Sympathetic chain and ganglia
Peripheral nerves
•
CNS alterations are secondary to axonal degeneration and affect
↘ Anterior horn cells in spinal grey matter
Neurons of motor cranial nerves nuclei in brainstem

Antecedent Events
C.Jujeni accounts for 1/3 of GBS because of mimicry between gangliosides and
lipopolysaccharides of the bacteria
Clinical features
1. AIDP
•
Infection (GIT: Campylobacter, Respiratory: Mycoplasma) within 2 weeks of
onset
•
Weakness(lower extremities then ascend up to the trunk then upper limbs and
bulbar weakness.
–
May start in the arms and move downward
–
May begin in the arms and legs at the same time
–
May occur in the nerves of the head only
–
In mild cases, weakness or paralysis may not occur

•
This weakness is symmytrical ( minor sides differences may occure), proximal
and distal
•
9% is asymmetrical
•
progress slowly over days or weeks Or abrupt and rapid
•
Child becomes irritable .
•
Parasthesia may occur, 89% pain accompany weakness
•
50% bulbar involvement .
•
Facial nerve involved. also VI,III,XII,IX,X
•
Some show viral meningitis or meningoencephalitis.
•
Papillodema ( unexplained pathogenesis )
•
Respiratory muscules : reduced vital capacity CO
2
retention even in
absence of respiratory symptoms
Features required for diagnosis
1. Progressive motor weakness of more than one limb.
2. Areflexia or hyporeflexia (loss of ankle jerks and diminished knee and biceps
reflexes will suffice if other features are consistent with the diagnosis.
Featrues supportive of diagnosis
1. Progression :weakness may develop rapidly but cease to progress after 4wk .
Roughly 50%will plateau within 2 wks , 80% by 3wks,and 90%by 4wks.
2. Relative symmetry .
3. Mild sensory symptoms and signs.
4. Cranial nerve involvements like facial weakness develops in about 50% of
patients.
5. Autonomic dysfunction.
6. Absence of fever at the onset of neurological symptoms.

7. Recovery without specific therapy, begins 2-4wks after progression ceases,
occasionally delayed for months.
Features casting doubt on the diagnosis
1. Marked persistent asymmetry in motor function.
2. persistent bowel or bladder dysfunction at onset of symptoms .
3. Discrete sensory level .
4. Progressive phase longer than 4wks .
5. CSF pleocytosis ( > 50wcc/mm3).
6. Complete ophthalmoplegia (internal or external).
Clinical Phases
Guillian-barre can be divided into five distinct clinical phases :
•
Phase 1- first 24 hr from presentation
•
Phase 2- disease progression
•
Phase 3- plateau phase
•
Phase 4- initial recovery
•
Phase 5- rehabilitation
Dx
1. Clinical
2. CSF
3. Electrophysiologic
4. MRI

Investigation
MRI of the brain and spinal cord
Should be considered in all patients ,usually done if :
1. The presentation is acute or rapidly progressive
2. There are predominantly sensory symptoms (including back pain)
3. There is predominant sphincter disturbance of presentation
4. There is a clear sensory or marked motor level
Lumber puncture
•
Elevated CSF protein without pleocytosis is a supportive diagnostic finding,
however the CSF may be normal within seven days of onset of symptoms
•
Protein level :elevated (>45mg/dl ) after the first week of symptoms , peak 4-5
wks
•
WCC <10/mm
3
, occasionally up to 50 mm
3
•
Glucose level normal
Neurophysiology
•
Normal nerve conduction studies in the first week does not exclude the
diagnosis of GBS
•
In AIDP nerve conduction impairment = conduction block , decrease compound
action potential amplitude
• Since the median duration of excretion of Campylobacter in stools of infected
persons is only 16 days and because of
the 1- to 3-week lag time between
infection and the onset of GBS,
many GBS patients with preceding
Campylobacter infection might
have falsely negative stool cultures.
• multiple
stool samples (or rectal swabs) should be obtained from GBS patients
immediately upon admission to the hospital, preferably 3 over
a 3-day period.

Other investigations
•
Full blood count, blood culture ( if pyrexial )
•
Urea and electrolytes ( hypokalemia )
•
Creatine kinase (myositis)
•
Chest x-ray , ECG
•
Abdominal x-ray ( palpable bladder , constipation )
Treatment
1. Admission
2. IVIG
3. Plasma exchange
4. Supportive treatment
5. Rehabilitation
• Symptomatic treatment is an essential part of the management of GBS.
• Children should admitted to the pediatric intensive care unit if they have one
or more of the following :
1. Flaccid tetraparesis
2. Severe rapidly progressive course
3. Reduced vital capacity at or below 20 ml/kg
4. Bulbar palsy with symptoms
5. Autonomic cardiovascular instability that is persistent hypertension or labile
blood pressure or arrhythmias.

Plasma exchange
•
Plasmapheresis has remained the gold standard treatment for GBS over the
last 20 years.
•
Should be used within 4 weeks of onset of neuropatic symptoms in non-
ambulatory patients
•
Should be used within 2 weeks of onset of neuropathic symptoms in
ambulatory patients
•
Plasmapheresis is generally safe in children who weigh 10 kg or more . A series
of exchange with a cumulative total of approximately 250 ml/kg volume
exchange or roughly a triple volume exchange .
•
Disadvantages of Plasmapheresis include its rare complications, such as sepsis,
risk of acquiring viral infections such as hepatitis and HIV.
IVIG treatment has advantages over plasmapheresis because
• it is easier to administer,
• has significantly fewer complications,
• and is more comfortable for the patient.
Side effects of IVIG
• expands the plasma volume so it must be administered with caution in
patients with congestive heart failure and renal insufficiency
• fever, myalgia, headache, nausea, and vomiting, but these "influenza-like"
symptoms are self-limiting.
• aseptic meningitis, neutropenia, and hypertension
• Anaphylaxis
• Thromboembolic events
• risk of serious hepatitis C infection transmission has been reduced

•
IVIG should be used within 2 weeks
•
Corticosteroid not recommended
•
Sequential treatment with PE then IVIG not recommended
•
PE & IVIG recommended for the severe disease
Pain management
Pain of discomfort is present in 50-80%of children with GBS at the time of
presentation .
Managing pain by:
•
Opioids
•
Non steroidal anti-inflammatory drugs ( ibuprofen)
•
Anti-epileptic drugs ( carbamazepine, gabapentine)
•
Tricyclic antiderpessants (amitryptine)
Prevention of pain:
•
Air matresses
•
Turning patients and carful positioning of limbs
•
Continuation of enteral feeding ,effective antacids as omeprazole
•
Preventing constipation
•
Prevent and treat urinary retention.
Supportive treatment directed to:
1. Hypertension , hypotension
2. Cardiac arrhythmia

3. Pulmonary embolism (Prophylaxis for deep venous thrombosis should be
provided because patients frequently are immobilized for many weeks).
4. Bladder and bowel
5. Psychological support
6. Nutrition , fluid , electrolytes
7. Pain
8. Skin
9. Cornea
10. Joints
11. Infection
12. communication
prognosis
40% → bed bound
15% → require ventilation
90-95% → complete recovery within 6-12months
Remainder ambulatory with minor residual deficit
4% → mortality rate
•
Antecedents C.jujeni infection correlates with poor Px
Causes of death :
1. Autonomic (bradycardia , tachycardia , hypertension)
2. Respiratory failure
3. Pulmonary embolism
4. Complication of ventilation
5. Cardiovascular collapse
Chronic relapsing or chronic unremitting (7%)
Features suggestive relapsing are:
1. Severely weak
2. Flaccid tetraplagia
3. Bulbar and respiratory muscle involvement
One or more relapses over 2mo.- years = CIDP
Congenital GBS
Weakness , areflexia , hypotonia .
CSF and electrophysical studies suggestive of GBS .
No treatment , gradual improvement.

Poliomyelitis
Polioviruses
•
RNA viruses, Picornaviridae family, enterovirus
•
3 genetically distinct serotypes
•
Spread from intestinal tract to CNS
•
90 – 95 % inapperant infections
•
Transmission: human is the only reservoir
•
Fecal – oral route
•
Isolated from stool for 2 weeks before paralysis to several weeks after onset of
symptoms
Pathogenesis
•
Wild type and vaccine strains
•
Gain host entry through GIT
•
Pass to the regional lymph nodes
•
Goes to the blood causing viremia
•
Wild type access the CNS through peripheral nerves
•
Incubation period 8-12 days
Clinical manifestions
Wild type follow one of the following courses :
1. 90 -95 % inapparent infection (no disease & no sequelae
2. 5% inabortive disease (influenza – like syndrome 1-2 wk after infection,
fever, malaise, anorexia , headache +/- vomiting) then recovery complete

3. Non-paralytic poliomyelitis
• 1%
• Signs of abortive type, fleeting paralysis of bladder and constipation.
• This is first phase (minor) then symptoms-free period then major phase
• O/E: nuchal rigidity, changes in the deep and superficial reflexes
(impending paralysis). No sensory defects
4. Paralytic poliomyelitis
• 0.1%
• Spinal type : major phase , sensory (paresthesia, hypersthesia), motor(
fasiculation and spasms) progress to
• Asymmetric paralysis of one leg, then 1 arm
• DTR initially active then diminished and absent
• Variable course: some progress, some recover
5. Bulbar type
• +/- spinal cord involvement
• Nasal voice or cry
• Difficulty in swallowing
• Accumulated pharyngeal secretion
• Absence of effective coughing
• Nasal regurgitation
• Deviation of palate, uvula, tongue
• Involvement of vital centers in the medulla
• Paralysis of vocal cords … hoarseness, aphonia
• Sometimes culminate into ascending paralysis (Landry type)
6. polioencephalitis
• Rare
• Seizures , coma , spastic paralsysis , increased reflexes
• Respiratory insufficiency
7. Paralytic polio with respiratory insufficiency
• Anxious expression
• Inability to speak without frequent pauses
• Increased RR
• Movement of ala nasi, accessory muscles
• Inability to cough or sniff

• Paradoxical abdominal movement
• Relative immobility of intercostal space
Diagnosis
•
Should be considered in any unimmunized or incompletely immunized child
with paralytic disease
•
Or any child with paralytic disease occurring 7-14 days after receiving the oral
vaccine
•
Stool : Isolate the virus in 2 stool specimen collected with 24 – 48 hr apart
•
Can isolate polio virus in 80 – 90 % in the first week and less than 20% within 3-
4 wk
•
CSF : normal in minor disease
•
Cells 20 -200 / mm3 initially then reduced
•
Protein : increase to reach 50 -100 mg/dl by 2
nd
week
•
CSF serology : seroconversion or 4 folds rise in antibody titers
Treatment
•
No specific treatment
•
Supportive: Limit progression, prevent skeletal deformities, prepare child and
family for prolonged treatment
Abortive poliomyelitis
•
Analgesics, sedatives
•
Attractive diet
•
Bed rest until temperature normalize
•
Avoidance of exertion for ensuing 2 wks

•
Careful neurologic and musculoskeletal examination
Non paralytic poliomyelitis
•
Same as abortive
•
Relief muscle tightness
•
Analgesics
•
Hot packs for 15-30 min every 2-4 hr
•
Hot tub baths
•
Firm bed
•
Footboard or splint to keep feet at right angle to legs
•
Later gentle physical therapy
Paralytic poliomyelitis
•
Hospitalization
•
Physical rest
•
Suitable body alignment to prevent deformity
•
Change position every 3-6 hr
•
Active and passive movement indicated as pain disappear
•
Moist hot packs
•
Opiates and sedatives
•
Treat constipation
•
Parasympathetic stimulant for bladder paralysis
•
Adequate dietary and fluid intake
•
Orthopedist, physiatrist should see them

Bulbar
•
Maintain airway
•
Avoid risk of inhalation
•
Gravity drainage of accumulated saliva
•
Nursed in lateral or semi-prone position
•
Aspirators with rigid tips for oral pharyngeal secretion or flexible catheter for
nasopharyngeal
•
Fluid and electrolytes equilibrium
•
Blood pressure taken at least twice
•
Impaired ventilation signs should be noticed to decide tracheostomy
Prognosis
•
Inapparent, abortive and aseptic meningitis = good outcome
•
Paralytic = depends on the extent and severity of CNS involvement, recovery
phase last 6 months
•
Severe bulbar = MR 60%
•
30 – 40 % of persons survived paralytic polio… may experience muscle pain and
exacerbation of exisiting weakness after 30 -40 yr
Prevention
•
Vaccination
•
Hygienic mearures
Thank you
