Pediatric hematology
Dr. Hayder Hadi Al-Musawi4/25/2016
1
4/25/2016
2Anemia's
Bleeding tendencyMalignancies
4/25/2016
3Pediatric anemia
introductionAnemia can be defined as a reduction in hemoglobin concentration, hematocrit, or number of red blood cells per cubic millimeter.
The lower limit of the normal range is set at two standard deviations below the mean for age and sex for the normal population.
4/25/2016
4classification
• I. Impaired red cell formation• 1. Deficiency
• Iron deficiency
• Folate deficiency
• Vitamin B12 deficiency
• Vitamin C deficiency
• Protein deficiency
• Vitamin B6 deficiency
• Thyroxine deficiency
4/25/2016
5
4/25/2016
62. The Bone marrow failure
A. Failure of a single cell line
B. Failure of all cell lines
C. Infiltration
4/25/2016
7II. Blood loss
III. Hemolytic anemia
A. Corpuscular
a. membrane defect: spherocytosis, elliptocytosis
b. enzymatic defects : G6PD, pyruvate kinase deficiency
c. hemoglobin defect:
1. Qualitative: sickle cell anemia
2. Quantitative: thalassemia
B. Extra corpuscular
i. Isoimmune: Rh incompatibilityii. Autoimmune
1. idiopathic: autoimmune hemolytic anemia
2. secondary : immunological disorder e.g. lupus
4/25/2016
8• IRON DEFICIENCY ANEMIA
introduction
Is the most common hematological problem of infancy and childhood.Iron is required for hemoglobin (Hb) synthesis, enzyme system and newly regenerated tissue
4/25/2016
9
Iron metabolism-1
The body of a newborn infant contains about 0.5g of iron, whereas the adult content is estimated at 5g.Accordingly, to maintain positive iron balance in childhood, about 1mg of iron must be absorbed each day.
Factors affect absorption and requirement of iron include:
• Amount:
because absorption of dietary iron is assumed to be about 10%, a diet containing 8–10mg of iron daily is necessary for optimal nutrition.
4/25/2016
10Iron metabolism-2
2) Type:
Iron is absorbed two to three times more efficiently from human milk than from cow's milk.
3) Age:
Infants breast-fed exclusively should receive iron supplementation from 4 mo of age. Adolescents are also susceptible to iron deficiency .
4/25/2016
11
Iron metabolism-3
4) Dietary factors:enhance iron absorption
• ascorbic acid
• meat
4/25/2016
12
Etiology-1
• Deficient intake:• Low birth weight and unusual perinatal
• hemorrhage:
• Increased demand: as in LBW, premature babies.
4/25/2016
13
Etiology-2
• blood loss :• Chronic diarrhea may be associated with unrecognized blood loss.
• Exchange transfusion:
• Intense exercise conditioning:
• Impaired absorption: malabsorption syndromes or celiac disease.
4/25/2016
14Clinical manifestations-1
Pallor is the most important clue to IDA.Blue sclera are also common, although also found in normal infants.
Irritability.
Pagophagia, the desire to ingest unusual substances such as ice or dirt, may be present.
Anorexia
Splenomegaly and stroke, rarely.
4/25/2016
15
Clinical manifestations-2
Neurologic and intellectual function, such as changes in attention span, alertness, and learning difficulties in both infants and adolescents.
4/25/2016
16Laboratory findings-1
• In progressive iron deficiency, a sequence of biochemical and hematologic events may occur :• First, the tissue iron stores represented by bone marrow hemosiderin disappear. The level of serum ferritin,decreased. Next,
• Serum iron level decreases, the iron-binding capacity of the serum increases, and the percent saturation decreases.
• Transferin saturation = S. iron <15% in IDA
• TIBC
4/25/2016
17
Laboratory findings-2
• Hematological changes: blood film:• As the deficiency progresses, the RBCs become smaller than normal (microcytosis) and their hemoglobin content decreases (hypochromia).
• The morphologic characteristics of RBCs are best quantified by the determination of mean corpuscular hemoglobin (MCH) and mean corpuscular volume (MCV).
• With increasing deficiency, the RBCs become deformed and misshaped and present characteristic poikilocytosis, and increased RBC distribution width (RDW).
4/25/2016
18
Laboratory findings-3
• The bone marrow is:
• Hypercellular, with erythroid hyperplasia.
• Occult blood can be detected in the stools of about one third of cases.
4/25/201619
Differential diagnosis
• Lead poisoning• Thalassemia syndrome
• Anemia of chronic inflammation and infection
4/25/2016
20
Treatment
Oral iron: 6 mg/Kg/ day of elemental iron in divided doses until normal Hb is achieved, then continue for 2-3 months to replace the iron store.Parenteral iron: intramuscular & not intravenous, but it has no advantages over the oral route.
Blood transfusion:
It is indicated only when the anemia is very severe.
4/25/2016
21
Hypochromic microcytic (iron deficiency)
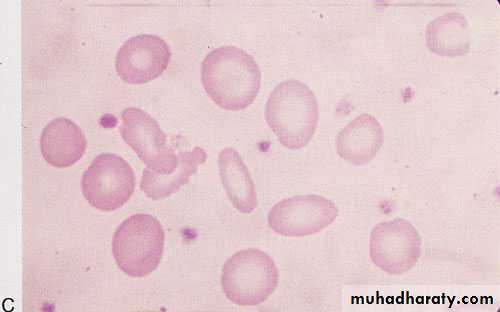
4/25/2016
224/25/2016
23MEGALOBALSTIC ANEMIAS
4/25/2016
24Megaloblastic morphology may be seen in a number of conditions; almost all cases in children result from a deficiency of folic acid, vitamin B12, or both.
Both substances are cofactors required in the synthesis of nucleoproteins.
FOLIC ACID DEFICIENCY
4/25/2016
25
Causes
• Primary : is rare• Inadequate dietary intake.
• Goat milk.
• Secondary: is more common, account for most cases:
• GIT: surgical removal or GIT disorders.• Increased requirement:
rapid growth
infections
4/25/2016
26
Clinical features
Peak incidence at the age of 4-7 months (slightly earlier than IDA).Mild anemia has been reported in very low birth weight infant and routine folic acid supplementation is required.
The usual clinical features of anemia.
4/25/2016
27
Laboratory findings
Blood smear (film) :
Macrocytic anemia ( high MCV).
The neutrophils are large, some with hypersegmented nuclei.
Decreased serum folic acid .
Levels of RBC folate are a better indicator of chronic deficiency.
Serum activity of lactic acid dehydrogenase (LDH) is markedly elevated.
The bone marrow is hypercellular because of erythroid hyperplasia. Megaloblastic changes are prominent.
4/25/2016
28
Treatment
folic acid may be administered orally or parenterally for one month.
Because a hematologic response can be expected within 72hr, blood transfusions are indicated only when the anemia is severe or the child is very ill.
4/25/2016
29
VITAMIN B12 DEFICIENCY
Source: vitamin B12 is derived from cobalamin in food from animal sources.Metabolism:
The cobalamins are released in the acidity of the stomach and combine there with R proteins and intrinsic factor (IF), and are absorbed in the distal ileum via specific receptors.
4/25/2016
30
Causes
dietary deficiency is rare. It may occur in cases of extreme dietary restriction (strict vegetarians: “vegans”) :Surgery or any disease involving the stomach or terminal ileum,
Juvenile Pernicious anemia:
Rare autosomal recessive disorder results from an inability to secrete gastric IF or secretion of a functionally abnormal IF.
The symptoms of juvenile pernicious anemia become prominent at 9mo–11yr of age. This interval is consistent with exhaustion of the stores of vitamin B12 acquired in utero.
Lack of secretion of IF by the stomach.
4/25/2016
31Clinical features
Signs and symptoms of anemia.
Sub acute dorsolateral degeneration of spinal cord (rare in children).
Neurologic manifestations include ataxia, paresthesias, hyporeflexia, Babinski responses, clonus, and coma.Glossitis (common in children).
4/25/201632
Laboratory findings-1
Peripheral smear (CBC) showsMacrocytic RBCs.
Neutrophil is large and hypersegmented
Serum B12 levels are decreased.
Serum LDH activity is markedly increased.
Excessive excretion of methylmalonic acid in the urine is a reliable and sensitive index of vitamin B12 deficiency.
4/25/2016
33
Laboratory findings-2
Absorption of vitamin B12 is usually assessed by :
Schilling test
Vitamin B12 and / or IF are given to confirm the diagnosis.
4/25/2016
34
Treatment
A prompt hematologic response follows parenteral administration of vitamin B12 (1mg).Oral therapy is not generally advisable owing to uncertainty of absorption.
4/25/2016
35
Morphologic abnormalities of the red blood cell. A, Normal. B, Macrocytes (folic acid or vitamin B12 deficiency).
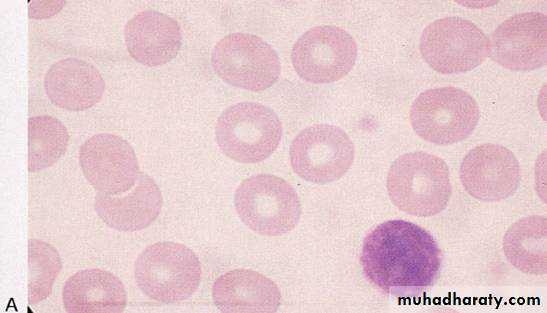
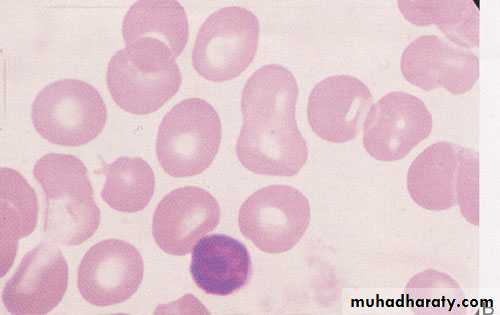
4/25/2016
364/25/2016
37HEMOLYTIC ANEMIAS
4/25/2016
38
Hemolysis is defined as the premature destruction of red blood cells (RBCs).
During hemolysis, RBC survival is shortened, and increased marrow activity results in an increased reticulocyte (new RBC) count.
the reticulocyte index is 1% normally.
Indirect bilirubin is increased leading to jaundice.
CBC : destructed,fragmented & misshaped RBCs (poikilocytosis). Hb levels are decreased.
Decreased serum haptoglobin levels.
Urine examination shows Hb in urine (hemoglobinurea) and appearance of urobilinogen.
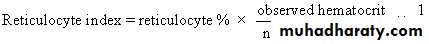
4/25/2016
39ENZYMETIC DEFECTS
4/25/2016
40Glucose 6 Phosphate Dehydrogenase Deficiencies
• Is the most important disease of the hexose monophosphate pathway.
• This is an example of X-linked recessive disease.
• It is responsible for two clinical syndromes:
• An acute episodic hemolytic anemia.
• Chronic hemolytic anemia.
4/25/2016
41
Clinical manifestations 1
• In the usual pattern of G6PD deficiency, symptoms develop 24–48hr after a patient has ingested a substance that has oxidant properties.
Drugs that have these properties include aspirin, sulfonamides, and antimalarials such as primaquine.
In some patients, ingestion of fava beans, may also produce an acute and severe hemolytic syndrome called favism.
Infection and fever are trigger factors.
4/25/2016
42Clinical manifestations 2
hemoglobinuria and jaundice may occur, and the hemoglobin concentration may fall precipitously with the resultant of life threatening condition.
Neonatal hyperbilirubinemia and potential kernicterus.
4/25/2016
43Laboratory findings
The onset of acute hemolysis results in a progressive fall in hemoglobin (Hb).
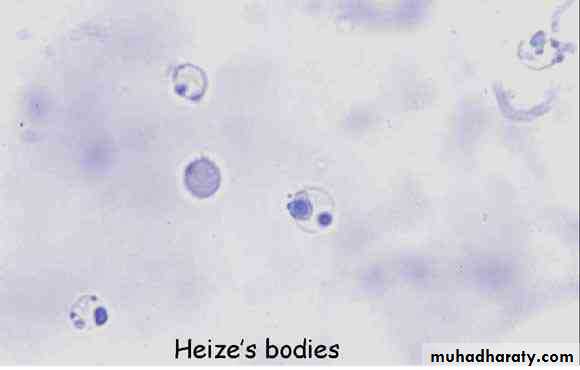
Blood film: unstained RBCs reveal Heinz bodies (precipitated hemoglobin). They are not seen after the first 3–4 days of illness. The blood film reveals a few fragmented RBCs.
4/25/2016
44
4/25/2016
45Diagnosis
• Demonstration of a reduced G6PD activity in RBCs. By direct measurement.
• The young RBCs (reticulocytes) have significantly higher enzyme activity than do older cells. Testing may therefore have to be deferred for 8-12 weeks before a diagnostically low level of enzyme can be shown.
• G6PD variants also can be detected by electrophoretic analysis.
4/25/2016
46Prevention and treatment
Prevention of hemolysis constitutes the most important therapeutic measure.
Avoidance of oxidants in known patients.
When hemolysis occurs, supportive therapy may require I/V fluids and blood transfusions, although recovery is the rule when the oxidant agent is removed.
4/25/2016
47STRUCTURAL DEFECTS
4/25/2016
48
HEREDITARY SPHEROCYTOSIS
Usually is transmitted as an autosomal dominant and, less frequently, as an autosomal recessive disorder, but it may occur because of mutations.
4/25/2016
49Clinical manifestations 1
Neonates: Hereditary spherocytosis may be a cause of hemolytic disease in the newborn and may present with anemia and hyperbilirubinemia sufficiently severe to require phototherapy or exchange transfusions.
Infant and older children: The severity is variable, but may have severe anemia with pallor, jaundice, fatigue, and exercise intolerance.
4/25/2016
50Clinical manifestations 2
Examination: splenomegaly & gallstones.
Aplastic crises: which is caused by a viral infection, called parvovirus B19 virus, and also hypoplastic crises are associated with various other infections..
4/25/2016
51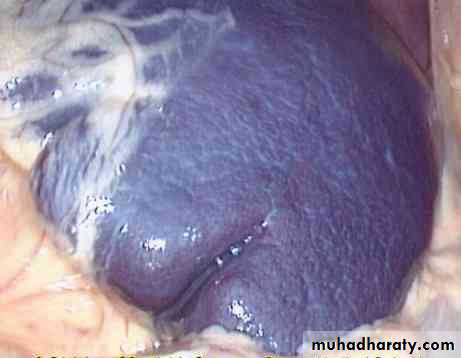
4/25/2016
52Laboratory findings
CBC: (complete blood count)
Spherocytes usually account for more than 15–20% of the cells when hemolytic anemia is present.
Erythroid hyperplasia is evident in the bone marrow aspirate or biopsy.
osmotic fragility test.
Auto hemolysis test.
gel electrophoresis: to demonstrate RBC membrane defect.
4/25/2016
53Treatment
splenectomy eliminates most of the hemolysis associated with this disorder.
splenectomy is not preferred before 5 years of age,
otherwise, patients should receive hepatitis B vaccination
and regular benzathin penecillin injections on monthly basis,
or penecillin V orally on daily basis.
this is due to loss of splenic immunologic function against
some viruses & encapsulated bacteria, such as pneumococci,meningococci, and Haemophilus influenzae type b.
Folic acid, should be administered to prevent secondary folic acid deficiency.
THALASSEMIA SYNDROMES
Introduction
The thalassemias are a heterogeneous group of heritable hypochromic anemias of various degrees of severity.Underlying genetic defects include total or partial deletions of globin chain genes.
Introduction 2
Important types of thalassemia are :α- thalassemia; which may lead to hydrops fetalis in sever cases.
β- thalassemia :
• thalassemia minor (trait).
• b) thalassemia major.
• c) thalassemia intermedia; which is in between the above.
HOMOZYGOUS β – THALASSEMIATHALASSEMIA MAJOR: COOLY`s ANEMIA
Clinical manifestationsPallor, hemosiderosis, and jaundice combine to produce a greenish-brown appearance color.
hypertrophy of erythropoietic tissue occurs in medullary and extramedullary locations. The bones become thin, and pathologic fractures may occur. Massive expansion of the marrow of the face and skull produces characteristic facies.
Hepatospleenomegaly.

Laboratory findings
CBC: The RBC morphologic abnormalities are extreme. Such as severe hypochromic and microcytic RBCs, with many bizarre, fragmented poikilocytes are present. The hemoglobin level falls progressively to lower than 5g/dL unless transfusions are given. .Icreased serum ferritin levels.
The diagnosis can be made by Hb – electrophoresis ( Hbf is increased).
Management
• Regular blood transfusion.• Iron Chelation therapy:
Like :
Desferal, which is given subcutaneousely for 12 hours 5 nights / week.
Exjade, which is an oral therapy.
D. Splenectomy.
E. Folic acid.
F. Bone marrow transfusion: the only curative Tx.
SICKLE CELL HEMOGLOBINOPATHIES
Introduction
Sickle hemoglobin (Hb S) differs from normal adult hemoglobin by a substitution of glutamic acid at the 6th position of its β chains by valine.
SICKLE CELL ANEMIA
Clinical manifestationsHemolytic anemia gradually develops over the 1st 2 – 4 months of life.
Acute sickle dactylitis (hand-foot syndrome),may occur during 1st year of life.
Crises
• Vaso occlusive crises• Acute painful episodes represent the most frequent and prominent manifestation of sickle cell disease.
• Intercurrent illnesses, fever, hypoxia, and acidosis, are trigger factors for these crises.
Splenic infarcts causing pain and “autosplenectomy.”
Pulmonary infarctions leading to chest pain and acute chest syndrome.
Strokes.
Ischemic damage may also affect any organ.
2. Sequestration crises
acute splenic sequestration• A plastic crisis
• with parvovirus B19 infection.
• Hyper hemolytic crises:
• sickle cell anemia and G6PD deficiency.
Laboratory findings
CBC: hemoglobin concentrations usually range from 5–9g/dL. The peripheral blood smear typically contains poikilocytes, and irreversibly sickled cells.
Diagnosis is made by Hb-electrophoresis ( HbS is increased).
Treatment
Preventive measures : to prevent serious illnesses by immunizations, prophylactic antibiotics, and prompt treatment of any infection.Painful crises : treated by analgesics and blood transfusions.
Splenectomy for repeated splenic sequestration crises.
Stimulation of Hbƒ synthesis : using drugs like hydroxyurea.
Bone marrow transplantation : curative measure.
4/25/2016
70thank you
