AnticancerAntimetaboliteslec 3 & 4
ByNohad A AlOmari
Most antimetabolites are effective cancer chemotherapeutic agents via interaction with the biosynthesis of nucleic acids. Therefore, several of the useful drugs used in antimetabolite therapy are purines, pyrimidines, folates, and related compounds.
The antimetabolite drugs may exert their effects by several individual mechanisms involving enzyme inhibition at active, allosteric, or related sites. Most of these targeted enzymes and processes are involved in the regulatory steps of cell division and cell/tissue growth. Often the administered drug is actually a prodrug form of an antimetabolite and requires activation in vivo to yield the active inhibitor.
The purine and pyrimidine antimetabolites are often compounds incorporated into nucleic acids and the nucleic acid polymers (DNA, RNA, etc.).
The antifolates are compounds designed to interact at cofactor sites for enzymes involved in the biosynthesis of nucleic acid bases. The biosynthesis of these nucleic acid bases depend heavily on the availability of folate cofactors, hence antimetabolites of the folates find utility as antineoplastic agents.
Pyrimidine DrugsThe anticancer drugs based on pyrimidine structure are shown in Figure 10.8.
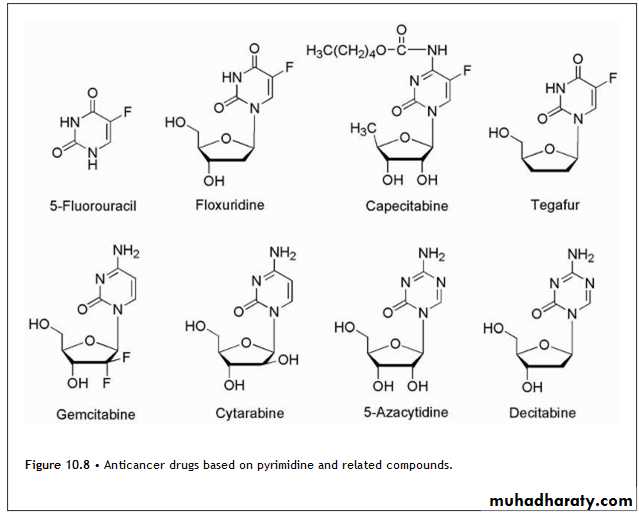
Modification of the pyrimidine ring has also been explored for the development of potential anticancer drugs based on antimetabolite theory.
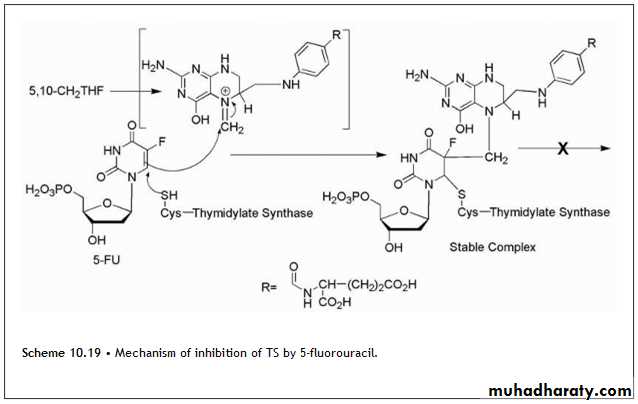
5-Fluorouracil is activated by conversion to the corresponding nucleotide species, 5-fluoro-2-deoxyuridylic acid .The resulting 5-fluoro-2′-deoxyuridylic acid is a powerful inhibitor of thymidylate synthetase, the enzyme that converts 2′-deoxyuridylic acid to thymidylic acid. In the inhibiting reaction, the sulfhydryl group of TS adds via conjugate addition to the 6-position of the fluorouracil moiety (Scheme 10.19), So the chemical mechanism of inhibition of TS by 5-fluorouracil is shown in Scheme 10.19.
Products5-FLUOROURACIL (5-FU, EFUDEX, aADRUCIL, FLUOROPLEX)CAPECITABINE (XELODA)
other mechanisms include that 5-FU in DNA likely serves as substrate for the editing and repair enzymes involved in DNA processing for cell division and tissue growth.Attempts at chemical modification of 5-FU to protect from catabolic events have produced several prodrug forms, which are converted via in vivo metabolic and/or chemical transformation to the parent drug 5-FU. Figure 10.8 shows the structure of the more common prodrug forms of 5-FU.
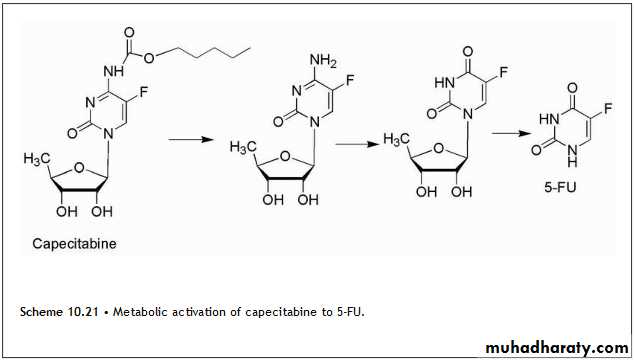
The carbamate derivative of 5′-deoxy-5-fluorocytidine is known as capecitabine, and it is converted to 5-FU through a series of activation steps. The activation sequence is shown in Scheme 10.21.
The initial step is carbamate hydrolysis followed by deamination, then hydrolysis of the sugar moiety to yield 5-FU. Some of these activation steps take place at a higher rate in tumor tissue leading to selective accumulation in those cells. The last step in the sequence shown in Scheme 10.21 is catalyzed by phosphorolases, and these enzymes occur in higher levels in colorectal tumors.
Despite this complex activation process, capecitabine still exhibits some of the significant toxicities of 5-fluorouracil. The tetrahydrofuran derivative tegafur is slowly converted to 5-FU but requires quite high doses to reach therapeutic plasma concentrations. Esters of the N-hydroxymethyl derivative of tegafur show greater anticancer activity than tegafur.(HW???)
Gemcitabine is the result of fluorination of the 2′-position of the sugar moiety. Gemcitabine is the 2′,2′-difluoro deoxycytidine species and after its anabolism to diphosphate and triphosphate metabolites, it inhibits ribonucleotide reductase and competes with 2′-deoxycytidine triphosphate for incorporation into DNA. The mechanism of action for gemcitabine includes alteration of the rate of incorporation into DNA as well as the rate of DNA processing and repair.
Purine DrugsThe anticancer drugs based on purine structure are shown in Figure 10.9.
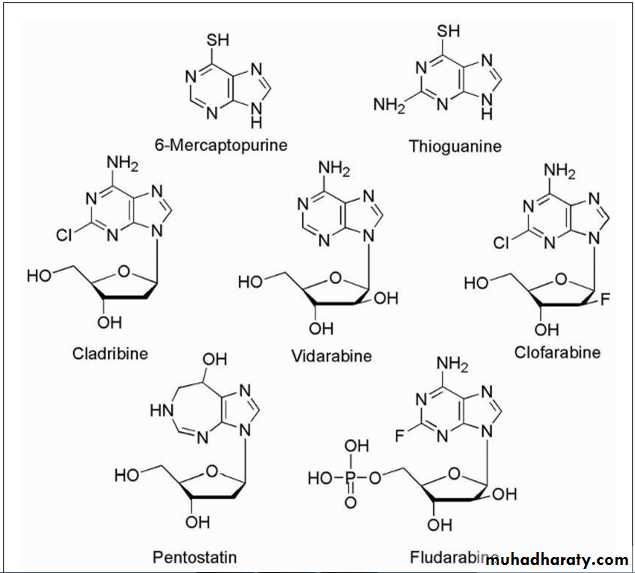
The antineoplastic activity of these purines as well as most antimetabolites depends on the relative rates of enzymatic activation and inactivation of these compounds in various tissues and cells. Drug resistance in certain cell lines may be caused by lower activity of activating enzymes or higher activity of catabolic enzymes.
The design of antimetabolites based on purine structure began with isosteric thiol/sulfhydryl group to replace the 6-hydroxyl group of hypoxanthine and guanine.
6-mercaptopurine (6-MP).
This purine requires bioactivation to its ribonucleotide, 6-thioinosinate (6-MPMP), by the enzyme HGPRT. The resulting nucleotide (Scheme 10.22) is a potent inhibitor of an early step in basic purine biosynthesis, the major antineoplastic action of 6-MP appears to be related to the inhibition of purine biosynthesis.
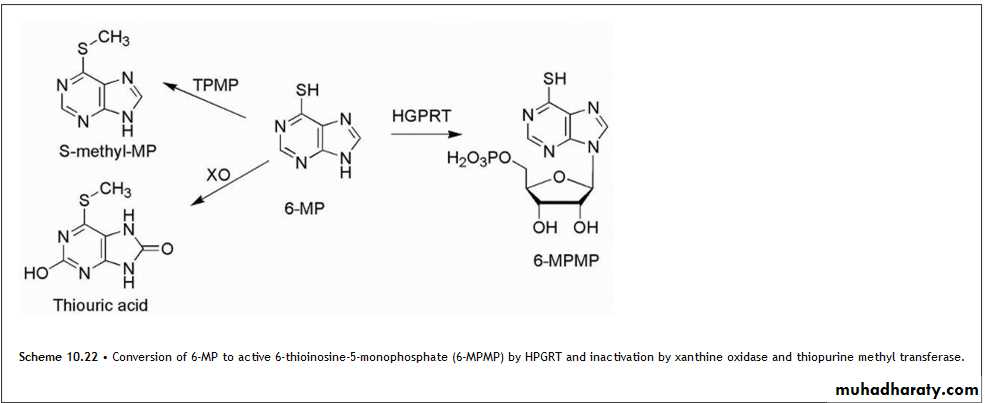
Thioguanine (6-TG) is the 6-mercapto analog of guanine, analogous to 6-MP. Thioguanine is converted into its ribonucleotide by the same enzyme that acts on 6-mercaptopurine. It is converted further into the diphosphates and triphosphates. These species inhibit most of the same enzymes that are inhibited by 6-mercaptopurine.
Thioguanine is also incorporated into RNA, and its 2′-deoxy metabolite is incorporated into DNA. The incorporation into RNA and DNA and the subsequent disruption of these polymers may account for a greater portion of the antineoplastic activity of thioguanine compared with 6-MP.
FolatesFolic acid and the structures of the major antifolate anticancer drugs are shown in Figure 10.10.
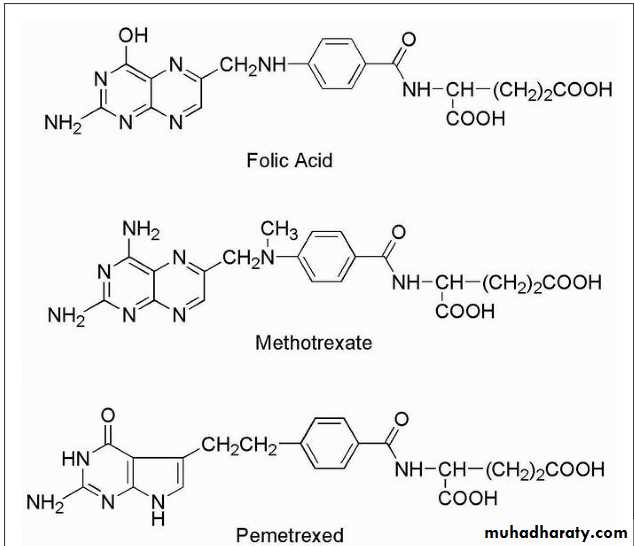
Methotrexate is the classic antimetabolite of folic acid structurally derived by N-methylation of the para-aminobenzoic acid residue (PABA) and replacement of a pteridine hydroxyl by the bioisosteric amino group.
The conversion of —OH to —NH2 increases the basicity and yields greater enzyme affinity. This drug competitively inhibits the binding of the substrate folic acid to the enzyme DHFR, resulting in reductions in the synthesis of nucleic acid bases, perhaps most importantly, the conversion of uridylate to thymidylate as catalyzed by thymidylate synthetase. In addition, purine synthesis is inhibited because the N-10-formyl tetrahydrofolic acid is a formyl donor involved in purine synthesis.
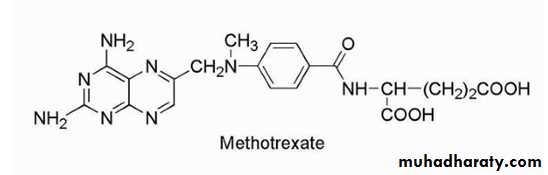
FLOXURIDINE
(FLUORODEOXYURIDINE, FUDR)The drug is available as a 500-mg vial of lyophilized powder. The drug is used to treat metastatic GI adenocarcinoma. The mechanism of action of this fluoropyrimidine deoxynucleoside analog involves metabolic conversion to 5-fluorouracil (5-FU) metabolites resulting in inhibition of TS thus disrupting DNA synthesis, function, and repair.
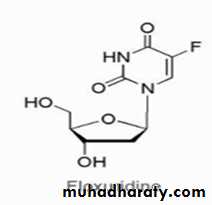
GEMCITABINE (DFDC, GEMZAR)
The drug is available as the hydrochloride salt in 200- and 1,000-mg lyophilized single-dose vials for IV use. Gemcitabine is used to treat bladder cancer, breast cancer pancreatic cancer. The mechanism of action of this fluorine-substituted deoxycytidine analog involves inhibition of DNA synthesis and function via DNA chain termination. The triphosphate metabolite is incorporated into DNA inhibiting several DNA polymerases and incorporated into RNA inhibiting proper function of mRNA.Resistance can occur because of decreased expression of the activation enzyme deoxycytidine kinase or decreased drug transport as well as increased expression of catabolic enzymes.
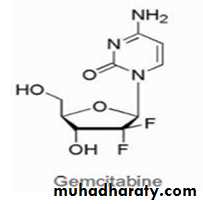
Cladribine is used for chronic lymphocytic
leukemia, hairy cell leukemia, andnon-Hodgkin's lymphoma.
The mechanism of action of this purine deoxyadenosine analog involves incorporation into DNA resulting in inhibition of DNA chain extension and inhibition of DNA synthesis and function. This incorporation into DNA occurs via the triphosphate metabolite active species. The 2-chloro group on the adenine ring produces resistance to breakdown by adenosine deaminase. Resistance to the anticancer effects can occur because of decreased expression of the activating enzyme or overexpression of the catabolic enzymes.
PENTOSTATIN (2′-DEOXYCOFORMYCIN, DCF, NIPENT)
The drug is available in 10-mg vials for IV use. The drug is used to treat leukemias such as hairy cell leukemia, chronic lymphocytic leukemia, and lymphoblastic leukemia. The mechanism of action involves inhibition of the enzyme adenosine deaminase yielding increased cellular levels of deoxyadenosine and deoxyadenosine triphosphate (dATP). The increased levels of dATP are cytotoxic to lymphocytes. Pentostatin is a fermentation product of Streptomyces antibioticus. Resistance appears to involve decreased cellular transport or increased expression of catabolic enzymes. Acid instability prevents oral administration, and the drug is only administered by IV.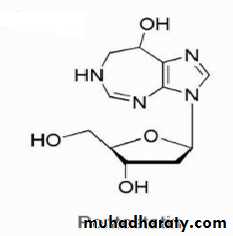
PEMETREXED (MTA, ALIMTA)
The drug is available in a 100-mg sterile vial for IV use. The drug appears to be effective against a range of tumors including mesothelioma, colorectal cancer, bladder cancer, and lung cancer. The mechanism of action involves inhibition of TS resulting in inhibition of thymidylate and DNA synthesis. This drug is a pyrrolopyrimidine analog of folate with antifolate activity. Resistance can occur by increased expression of TS, decreased binding affinity for TS, or decreased drug transport into cells.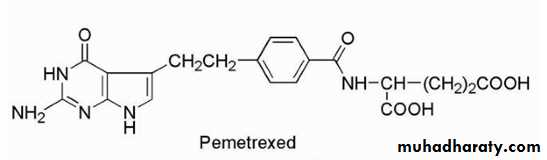
3. ANTIBIOTICS AND NATURAL PRODUCTS
3. ANTIBIOTICS AND NATURAL PRODUCTSA variety of the anticancer agents available today are derived from natural sources with several of these being obtained from microbial sources (antibiotics). Many of the antineoplastic antibiotics are produced by the soil fungus Streptomyces.
Both the antibiotic and natural product classes have multiple inhibitory effects on cell growth; however, they primarily act to disrupt DNA function and cell division.
There are several mechanisms by which these agents target DNA, including intercalation, alkylation, and strand breakage either directly or as a result of enzyme inhibition. Intercalation is a process by which a planar molecule of the appropriate size inserts itself between adjacent base pairs of DNA and in so doing, it causes a local unwinding that may disrupt the normal template function of DNA. Intercalation requires that the drug induce a cavity between base pairs so that insertion may occur.
The interaction of the intercalator and the adjacent base pairs occurs by the overlap of p-orbitals of the intercalator and the base pairs. The p-orbitals of the intercalation species are provided by a combination of aromatic and conjugated systems that impart the planarity required for intercalation.
Alternative modes of stabilization may occur through a combination of van der Waals interaction or hydrogen bonds.
The overall result of these interactions is to cause a local bend or kink in DNA resulting in a local shape distortion. This may produce several effects but is often associated with inhibition of normal DNA function.
By virtue of their ability to induce bends or kinks in DNA, intercalators may also result in inhibition of topoisomerase enzymes.
There are several natural products that are capable of disrupting the formation and function of the mitotic spindle.
These include the epipodophyllotoxins, the taxanes, and the vinca alkaloids.
The mitotic spindle forms during the M phase of the cell cycle and is responsible for moving the replicated DNA to opposite ends of the cell in preparation for cell division.During prophase, the cytoskeleton made of proteins, which gives the cell its shape, begins to breakdown providing the building blocks for the mitotic spindle. The cell then progresses to prometaphase, where the nuclear membrane begins to disappear and the spindle begins to form attaching the centrosomes to the kinetochores, which are proteins joining the two sister chromatids. Tension is then applied to the spindle so that during metaphase, the chromatids align near the center of the cell.
In anaphase, a further increase in tension separates the sister chromatids at the kinetochore, and they are pulled to opposite ends of the cell.
Ultimately, the microtubules will disintegrate and reform the cytoskeleton. The process is highly controlled and there are several proteins including “motor proteins,” which are involved in coordinating the process.
Actinomycin D
Is known as dactinomycin and contains identical pentapeptides bound through an amide linkage utilizing the amino group of L-threonine with carbonyls at positions 1 and 9. The pentapeptides namely L-threonine, D-valine, L-proline, sarcosine, and L-methylvaline form a lactone via the side chain hydroxyl of L-threonine and the carboxyl group of L-methylvaline (Fig. 10.11).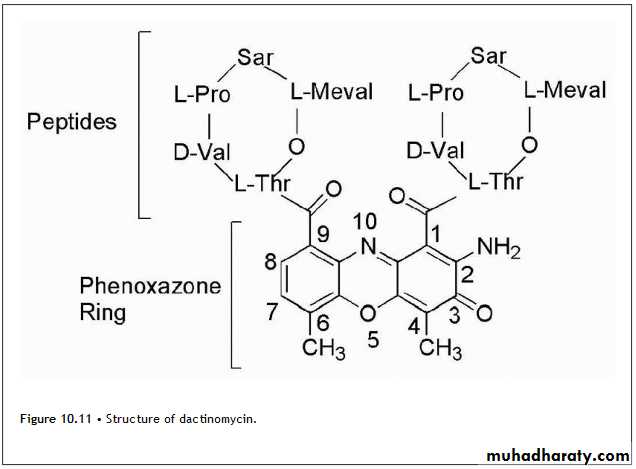
Anthracyclines
The anthracycline antibiotics (Fig. 10.13) are characterized by a planar oxidized anthracene nucleus fused to a cyclohexane ring that is subsequently connected via a glycosidic linkage to an amino sugar.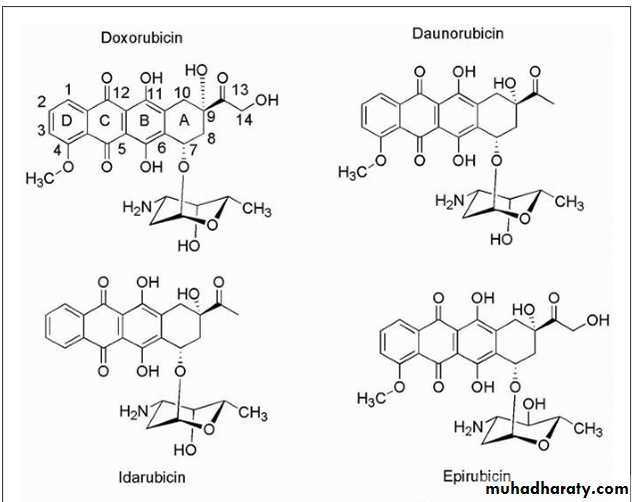
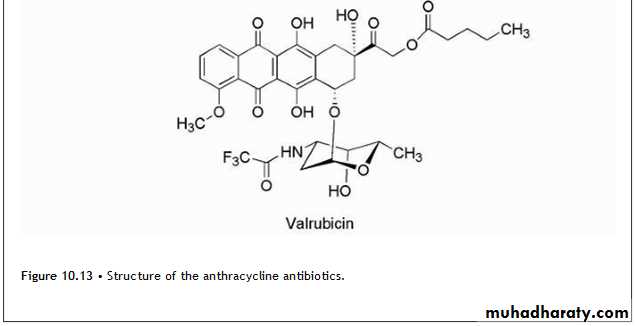
• The mechanism by which the anthracyclines exhibit their cytotoxic effects initially focused on the ability of the compounds to associate with DNA resulting from intercalation of their planar ring system reinforced by auxiliary binding of the amino sugar.
• Other mechanism involves intercalation followed by inhibition of topoisomerase II resulting in strand breakage leading to apoptosis.
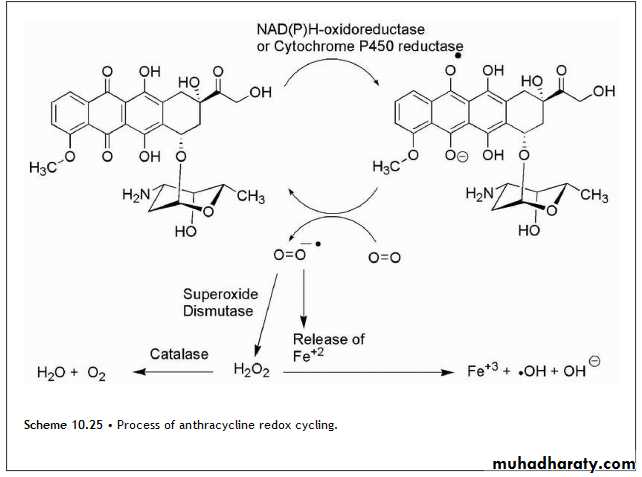
• Although the anthracyclines produce several adverse effects that are typical for antineoplastics, cardiotoxicity is a special concern with this class of agents. The associated cardiomyopathies and congestive heart failure (CHF) have been related to the ability of these compounds to undergo redox cycling Fig(10.25).
• Additional mechanisms of cardiotoxicity have been proposed, which involve the metabolic reduction of the anthracycline C-13 ketone to the alcohol.
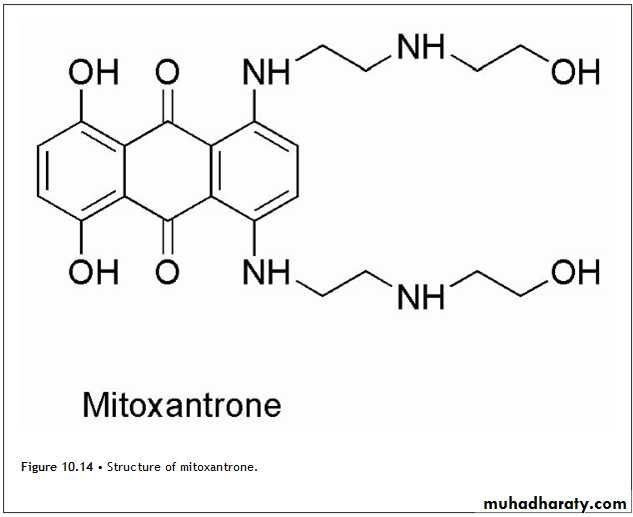
Mitoxantrone,
It is a derivative of a synthetic dye and is classified as an anthracenedione. (Figure 10.14.) Although mitoxantrone is a synthetic agent, it is included with the natural products because it is mechanistically similar to the anthracyclines.The mechanism involves intercalation by the chromophore, which is stabilized by the 2-[(2-aminoethyl)amino]ethanol side chain presumably as a result of an ionic interaction of the protonated amines with the phosphate backbone of DNA.
The formation of covalent adducts with DNA have also been demonstrated to occur in a manner similar to the anthracyclines.
In this case, however, other enzymes such as myeloperoxidase are responsible for the generation of formaldehyde.
Topoisomerase II is inhibited, and strand breakage occurs similar to that seen with the anthracyclines. In contrast to the anthracyclines, mitoxantrone is not a substrate for the reductase enzymes responsible for the conversion to the semiquinone so that ROS are not generated by this process. !!!!!
This lack of activation has been attributed to the presence of the side chains. This has the effect of reducing the cardiotoxicity but not completely eliminating it, and caution should be used especially in those patients with existing cardiovascular problems.
EpipodophyllotoxinsThe epipodophyllotoxins (Fig. 10.15) are semisynthetic derivatives of podophyllotoxin, which is isolated from the mayapple (mandrake) root and functions as an inhibitor of microtubule function.
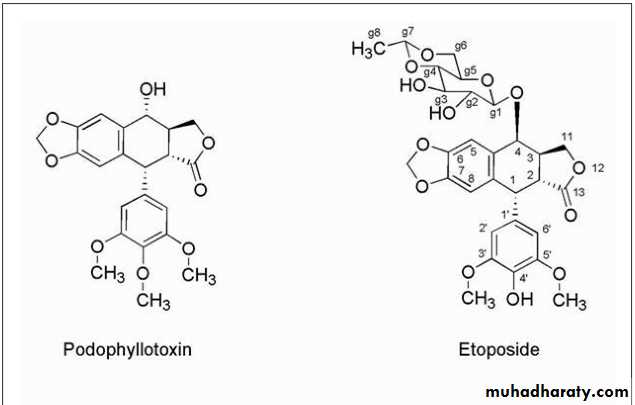
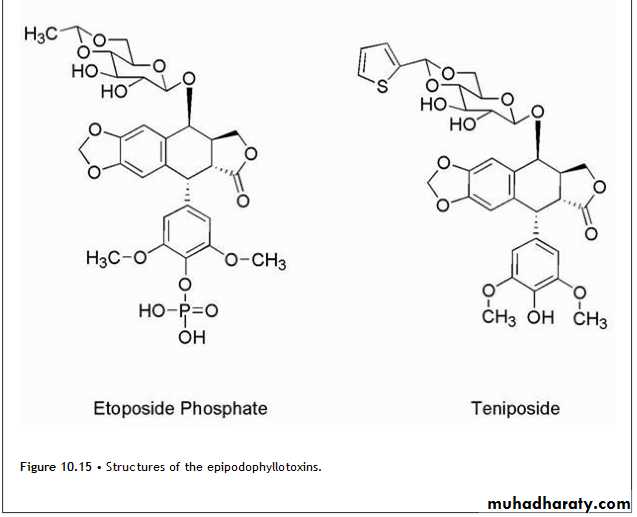
Chemical modification has led to compounds with a different mechanism of action, which involves inhibition of topoisomerase enzymes.
The change in mechanism was associated with removal of the 4′-methyl group of podophyllotoxin. Further alteration in podophyllotoxin involved the addition of the glycosidic portion of the molecules
Etoposide acts on topoisomerase II stabilizing the cleavable complex leading to single- and double-strand breaks. If enough breaks are initiated, apoptosis is activated.
Etoposide is believed to bind to topoisomerase II in the absence of DNA, because it shows little tendency to interact with DNA alone.
The etoposide-topoisomerase II complex then binds DNA, and strand cleavage occurs; however, the ligation step is inhibited.
Binding of the drug occurs near the site at which the cleaved phosphodiester bond is held by the enzyme.
One etoposide molecule stabilizes the cleavable complex of one chain, and therefore two etoposide molecules are necessary to mediate double-strand breaks.
The concentration of the agent will then determine whether single- or double-strand breaks occur. The agents are considered cell cycle specific and act in the late S and G2 phases of the cell cycle.
The glycosidic moiety of the epipodophyllotoxins, which is lacking in podophyllotoxin, is associated with converting these compounds from tublin binders to topoisomerase inhibitors.
There are several mechanisms by which cells become resistant to the epipodophyllotoxins including increased efflux by Pgp as seen for many of the other natural products.
Additionally, topoisomerase II levels may decrease or develop altered binding sites with lower affinity for these agents.
Increased DNA repair mechanisms may also decrease the effectiveness of these agents.
There are mechanisms by which double-strand breaks in DNA can be repaired. If for example a strand break occurs in the late portion of the S phase or in the G2 phase after DNA has been replicated, the sister chromatid can be utilized as a template to make the repair in a process known as homologous recombination without the loss of genetic material. However, if a sister chromatid is not available, then a process known as nonhomologus end joining may be utilized.Camptothecins
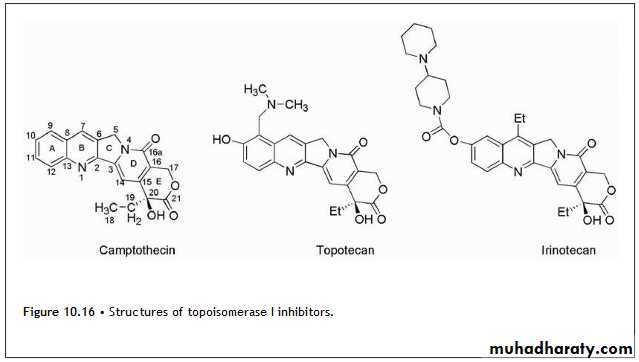
The camptothecins (Fig. 10.16) are inhibitors of topoisomerase I and are used clinically for the treatment of various cancers.
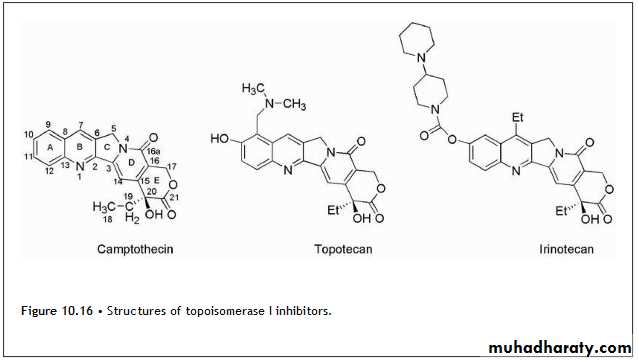
The lead drug for this class of agents was camptothecin, which was isolated from Camptotheca acuminata, an ornamental tree found in China.
The mechanism of antitumor action was inhibition of topoisomerase I. Subsequently, the incorporation of side chains containing basic amines led to the more water-soluble derivatives, topotecan and irinotecan (Fig. 10.16).
The camptothecin analogs bind to the enzyme DNA complex after strand cleavage has occurred, such that the planar structure of the drug can intercalate between DNA base pairs and then stabilize the cleavable complex. The binding site for this intercalation is only formed after the enzyme is bound to DNA, and once this site is occupied by the drug, it prevents the realignment necessary for resealing of the initial strand break.
In the case of topotecan, the results indicate that the planar portion of the drug intercalates between base pairs at the DNA cleavage site. In this process, the 5′-OH of the cleaved strand is shifted away from the phosphotyrosine bond formed between the enzyme and DNA. This shift prevents the 5′-OH from attacking the phosphotyrosine bond and resealing the strands after supercoiling has been removed. The result is strand breakage.
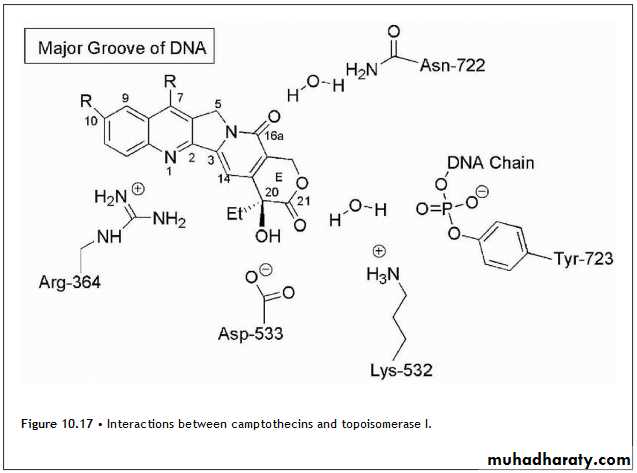
Other key interactions involve a direct hydrogen bond between Asp-533 of topoisomerase I and the 20-OH of topotecan.
There is a water-bridged hydrogen bond between the lactone carbonyl at C-21of the drug and the phosphotyrosine of the covalent topoisomerase I-DNA adduct.
Arg-364 is located in proximity to the quinoline nitrogen at N-1, although the interatomic distance was not close enough to allow for a specific interaction in the case of topotecan. Subsequent work with camptothecin, which binds with a slight twist of the planar ring system relative to topetecan, allowed for the formation of a hydrogen bond between Arg-364 and the N-1 nitrogen. The drugs orient such that C-7, C-9, and C-10 are directed into the major groove so that bulky substituent can be tolerated at these positions.
These interactions are summarized in Figure 10.17. The result of this binding is the formation of single-strand breaks most notably at replication forks that occur during the S phase. Strand breakage of the leading strand of DNA during replication results in double-strand breakage and ultimately leads to cell death.
Several mechanisms of resistance to the camptothecin analogs are known. Different neoplasms express different levels of topoisomerase I, and this is thought to explain the susceptibility of any specific cancer to these agents.
It is also possible for cells to decrease their expression of this enzyme or develop altered forms to which these compounds can no longer bind.
Increased DNA-repair enzymes may limit the damage to DNA, and these agents are susceptible to Pgp mediated efflux.
The agents have also been shown to activate nuclear factor-κB (NF-κB), which has an antiapoptotic effect so that strand breaks may not initiate apoptosis.
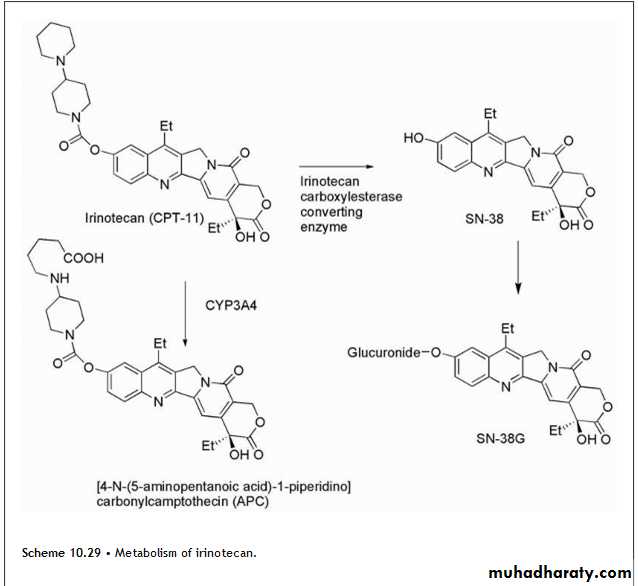
Irinotecan undergoes hydrolysis of its carbamate moiety by irinotecan-converting enzyme to give SN-38, which is 1,000 times more potent than the parent compound (Scheme 10.29). There is wide interpatient variability in the extent of this transformation, which may explain differential responses to the agent. Further metabolism involves the glucuronidation of the resulting phenolic function of SN-38 to give SN-38G, which is inactive.
An additional metabolite forms as a result of CYP3A4-mediated conversion to [4-N-(5-aminopentanoic acid)-1-piperidino]carbonylcamptothecin (APC), which is 100 times less active than SN-38. There is also the additional complication that the parent and metabolites may exist as the lactones or as the inactive hydroxy acid forms.