Motor neuron disease
Motor Neuron Disease MNDMND characterized by degeneration of:
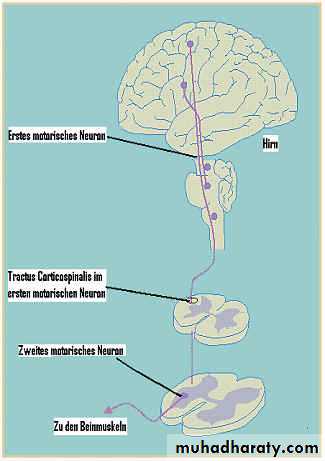
1- Anterior horn cells in the spinal cord.
2- Motor nuclei of the lower cranial nerves in the brainstem.3- pyramidal neurons in the motor cortex of the brain.
Etiology:-
• About 5% of cases are familial; autosomal dominant, the genetic defect lies on chromosome 21; the enzyme involved being a superoxide dismutase ((SODI))• About 95% of cases the possible causes include viral infection, trauma, exposure to toxin and electric shock but no evidence to support any of these
Clinical features:
- Usually after the age of 50 years.
- Very uncommon before the age of 30Years
- Affect males more than females
Classification1- Progressive bulbar palsy ((LMNL))
Occurs due to involvement of the motor nuclei of lower cranial nerves in the brainstem affecting the tongue, palate, and pharyngeal muscles leading to wasting and fasciculation of tongue with dysphagia and dysartheria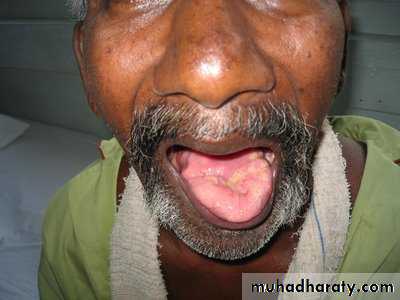
2- Pseudobulbar palsy: ((UMNL))
Occurs due to involvement of bilateral corticobulbar pathway causing contracted spastic tongue with dysphagia and dysartheria3- primary lateral sclerosis: ((UMNL))
This is pure upper motor ((corticospinal pathway )) deficit in the upper and lower limbs causing weakness, spasticity, exaggerated reflexes and extensor plantar responses
4- Progressive spinal muscular atrophy: ((LMNL))
Occurs due to degeneration of the anterior horn cells in the spinal cord; leading to weakness, wasting, fasciculation of the muscles in the upper and lower limbs with absent tendon reflexes
5- Amyotrophic lateral sclerosis:
A mixed upper and lower motor neuron deficit in the limbs and bulbar involvement leading toFatigability, muscle cramps, weakness, wasting and fasciculation of the muscles
Spasticity, exaggerated reflexes and extensor plantar responses- Bulbar and pseudobulbar palsy may follow eventually
Notes:
• External ocular muscles and sphincters usually remain intact• No objective sensory deficit
• No intellectual impairment in most cases
Investigations:
Electromyography ((EMG))Shows: denervation, fasciculation and some reduction in the amplitude of action potentials
* CSF examination; normal or slight elevation of protein
Management:
• Riluzole ((glutamate antagonist)) 100mg/day may reduce mortality and slow progression of amyotrophic lateral sclerosis
• Feeding via nasogastric tube may be required for severe dysphagia.
Prognosis:
Younger patients and those with early bulbar symptoms tend to show amore rapid course.Death within 3-5years of the onset of symptoms. Usually from respiratory infection and failure and complication of immobility.
Spinal Muscular Atrophy
• Prognosis• Features
• inheritance
• Onset
• Type
• poor
• Severe muscle wasting/ weakness
• Autosomal recessive
• Infancy
• Werdnig-hoffmann
• Slowly progressive disability
• Proximal weakness and wasting, EMG shows denervation
• Autosomal recessive
• Childhood, adolescence
• Kugelberg-welander
• Good, seldom disabling
• Distal weakness and wasting of hands and feet
• Autosomal dominant
• Early adult life
• Distal forms
• good
• Facial and bulbar weakness, proximal limb weakness, gynaecomastia
• X-linked
• Adult life, males only
• Bulbospinal
MYASTHENIA GRAVIS
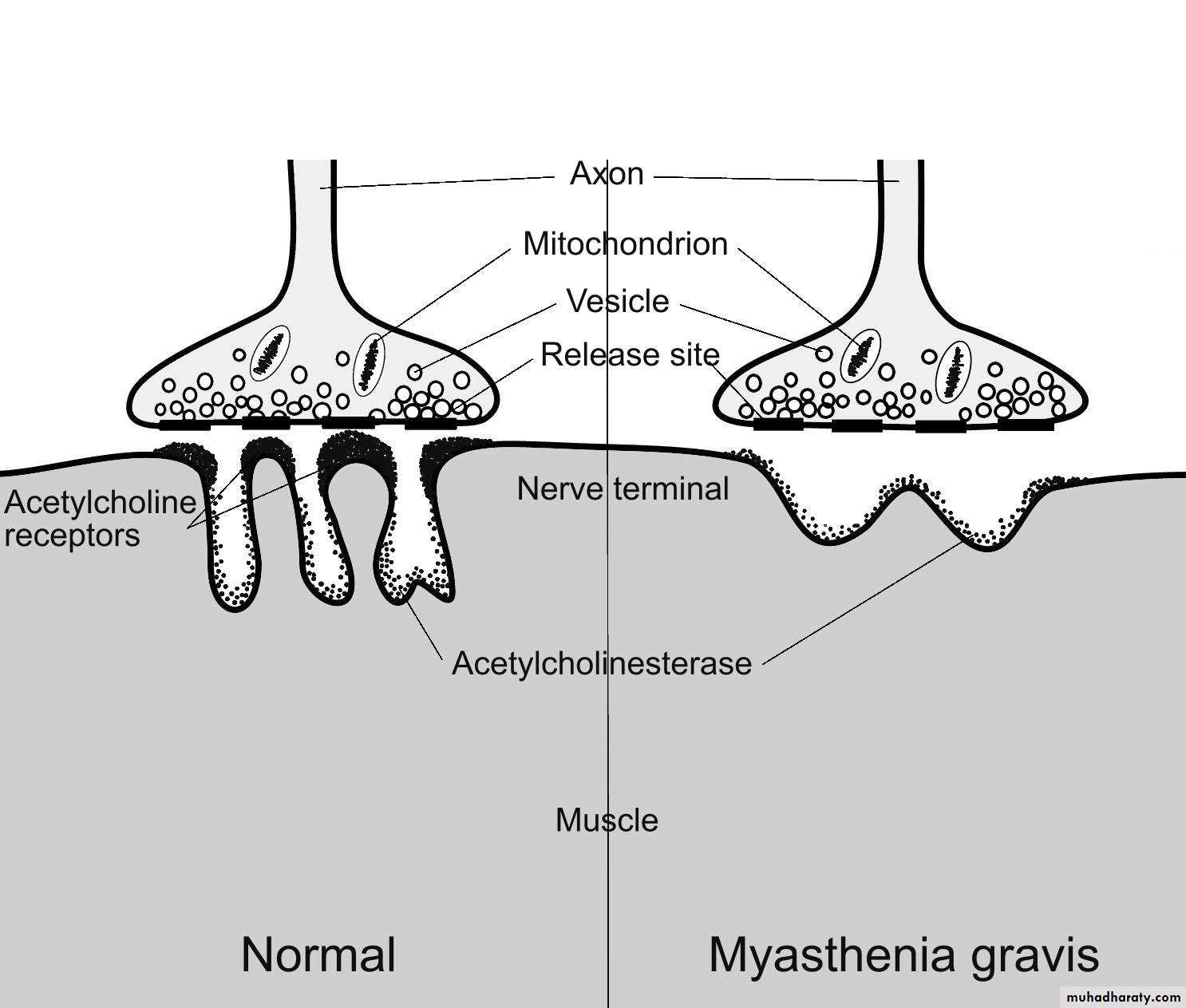
A disorder of the neuromuscular junction of the skeletal muscles characterized by fluctuating weakness and easy fatigability of the voluntary muscles ((muscle activity cannot be maintained , and initially powerful movement weaken rapidly))There is a predilection for the external ocular
muscle and certain other cranial muscleincluding the masticatory, facial, pharyngeal and laryngeal muscles.
Respiratory and limb muscles may also be affected.
Weakness is due to autoantibodies to acetylcholine receptors in the post junctional membrane of the neuromuscular junction . The result is decrease in the number of functioning acetylcholine receptors leading to the block of neuromuscular transmission.
Myasthenia gravis may be associated with thymic tumor , thyrotoxicosis , SLE , Rh arthritis.
Clinical features
- Can occur at any age but usually between age of 15 and 50 years .- More common in female than male.
- Insidious onset.
-Ptosis, diplopia, difficulty in chewing orswallowing and nasal speech, respiratory
difficulties or weakness of limbs.▪These symptoms often fluctuating in intensity during the day((diurnal variation )).
▪Spontaneous relapses and remissions my occur.
▪Exacerbation can occur due to infection, pregnancy and before menses.Symptoms may worsened by drugs
such as tetracycline,aminoglycosideciprofloxacin, propranolol,phenytoin,
lithium, procainamide, quinine.andquinedine so should be avoided in such patients
-No muscle atrophy-No reflex changes
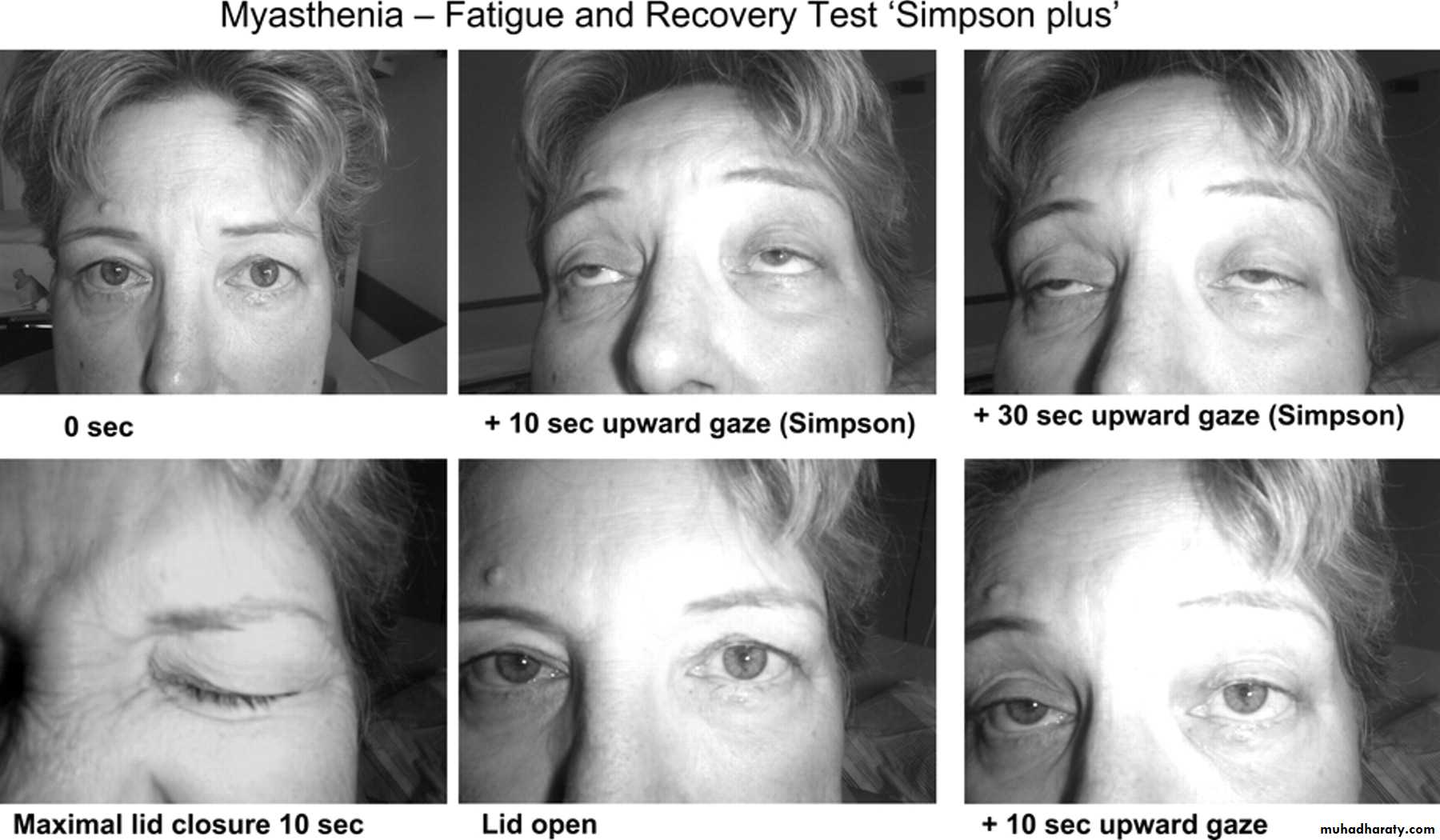
-No sensory changesConfirm the weakness and fatigability by
sustained up gaze for tow minutes, repeated knee bending
Diagnosis
-Clinical-Tensilon test: by using edrophonium IV in dose of 10mg (1ml)
-1.5mg of neostigmine 1M and atropine sulfate (0.6mg) should be available to counteract the muscarinic cholinergic side effect ((of neostigimine))
-EMG
-AChRA (Acetylcholine receptor antibody)-Anti-MuSK(muscle-specific kinase ) antibodies are found especially in AChRA-negative patient with prominent bulbar involvement .
-Anti-skeletal muscle antibodies , if positive suggest the presence of thymoma
-CXR and CT scan of chest for coexisting thymoma
Treatment
1-Anticholinestrase drugs▪Pyridostigmine is the mainstay of treatment given average 60mg orally, four times daily.
▪Neostigmine may use in rare instances by parental administration.
Over medication causing cholinergic crisis
which characterized by weakness unaffectedor enhanced by IV edrophonium , pallor ,
sweating , nausea , vomiting , salivation ,
colicky abdominal pain and miosis.
2-Thymectomy
Done for patient under 60years of age and those with weakness not restricted to the extra ocular muscles.3-Corticosteroids
Are indicated for patient who have responded poorly to anticholinesterase drugs and have thymectomy
The treatment should initiated in the hospital as the weakness may initially be exacerbated
4-Azathioprine
2-3mg/kg/day. used in patient with severe or progressive disease despite thymectomy and treatment with anticholenesterases and corticosteroids
5-Plasmaphersis
Used to achieve temporary improvement in patient deteriorating rapidly or in myasthenic crisis or prior to surgery
6-Intravenous immunoglobulin
Used to provide temporary benefit in circumstances similar to those in which plasmapheresis is usedPrognosis
Most patient can be managed successfully,the disease may have fatal outcome because
respiratory complication such as aspiration
pneumonia
Muscular dystrophy
Muscular dystrophies are a group ofinherited myophatic disorders characterized
by progressive degeneration of a group of
muscles without involvement of the
nervous system.
I-Duchenne׳s muscle dystrophy
▪The most common form of muscular dystrophy▪X - linked affect predominantly males
▪Begin by age of 3 years and by age of 12 years becomes wheel chair dependent ▪Toe walk , waddling gait , fall frequently and have difficulty keeping up with friends when playing running and jumping.
I-Duchenne׳s Muscle Dystrophy
▪The most common form of muscular dystrophy
▪X - linked affect predominantly males
▪Begin by age of 3 years and by age of 12 years becomes wheel chair dependent ▪Toe walk , waddling gait , fall frequently and have difficulty keeping up with friends when playing running and jumping.
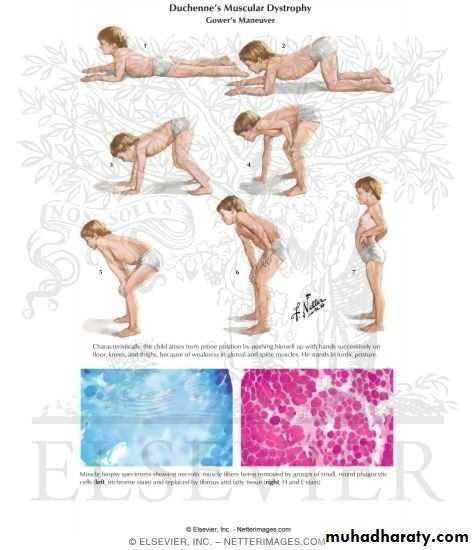
▪Weakness of the proximal muscle in the lower extremities > upper extremities ▪Gower׳s sign positive ((on getting up from the floor, the patient uses his hand to climb up himself))
▪Pseudohypertrophy of clave
▪Cardiomyopothy and mental retardation
▪Death in the third decade
Investigations
-Serum CK elevated between 20 and 100 times normal-EMG shows feature of myopathy
-Muscle biopsy shows muscle fiber of varying size, necrosis and replace by fat and connective tissue
-Definitive diagnosis established on the basis of dystrophin deficiency in a biopsy of muscle or mutation analysis on peripheral blood leukocytes.
-Screening for an associated cardiac abnormality ( cardiomyopathy or dysrhythmia ) is important .
Treatment
-Prednisolone in a dose 0.75 mg / kg / day, orally significantly slow progression of the disease for up to 3years.
-Physiotherapy and occupational therapy.
-Treatment of cardiac failure or arrhythmia.
-Treatment of respiratory complication .
II-Becker Dystrophy
•X-linked•It׳s average onset 11years and death of 40s
•The pattern of weakness similar to that of Duchenne׳s dystrophy
•Cardiac involvement less frequent than Duchenne's dystrophy