
1
Fifth stage
Pediatric
Lec. 4
.د
انور
12/4/2017
HEMOLYTIC ANEMIAS
Hemolysis : an increased rate of RBC destruction with a shortening of the normal life span
of the cell from the normal 120 days to as little as a few days in severe hemolysis. The
marrow can increase erythrocyte production 6-8 fold so mild degrees of hemolysis are not
associated with anemia.
Severe hemolysis can lead to a rapid and profound fall in Hb, and be life threatening. It is
suspected when polychromatic cells are seen on the blood film (reticulocytes).
It is usually associated with raised blood unconjugated bilirubin and splenomegaly .
Hemolysis can be caused by inherited or acquired disorders.
Diagnosis of the cause of the hemolysis is made according to the family history; clinical
features and red cell morphology that together will indicate what further laboratory tests
are required.
Marrow examination is generally unnecessary.
➢ The child with hemolysis may be pale with fluctuating jaundice (usually mild) and
splenomegaly.
➢ Pigment gallstones may complicate the disorder ,HA should always be excluded in a
child with stones.
➢ Aplastic crises may occur, usually precipitated by parvovirus infection which leads to
reticulocytopenia and anemia. Parvovirus infection typically produces severe anemia
sometimes requiring transfusion, and a modest thrombocytopenia and leucopenia.
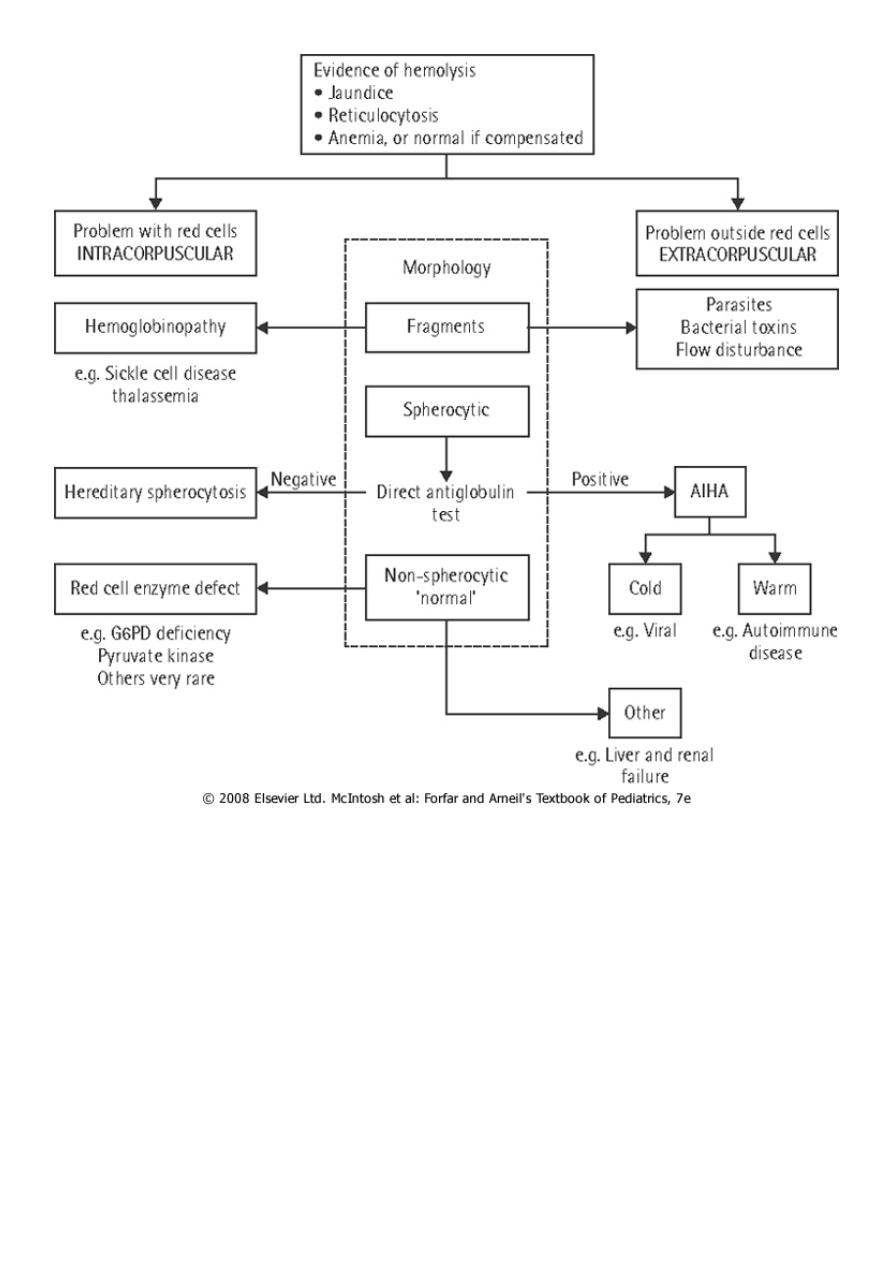
The major categories of HA are:
1. Immune mediated.
2. Membrane defect (Spherocytosis).
3. Enzyme defect (G6PD def).
4. Hb defect (SCA, Thalassemia).
2
3
Membrane defects
Hereditary spherocytosis (HS)
It is caused by a defect in the skeleton of the RBC membrane that generally affects the
spectrin component.
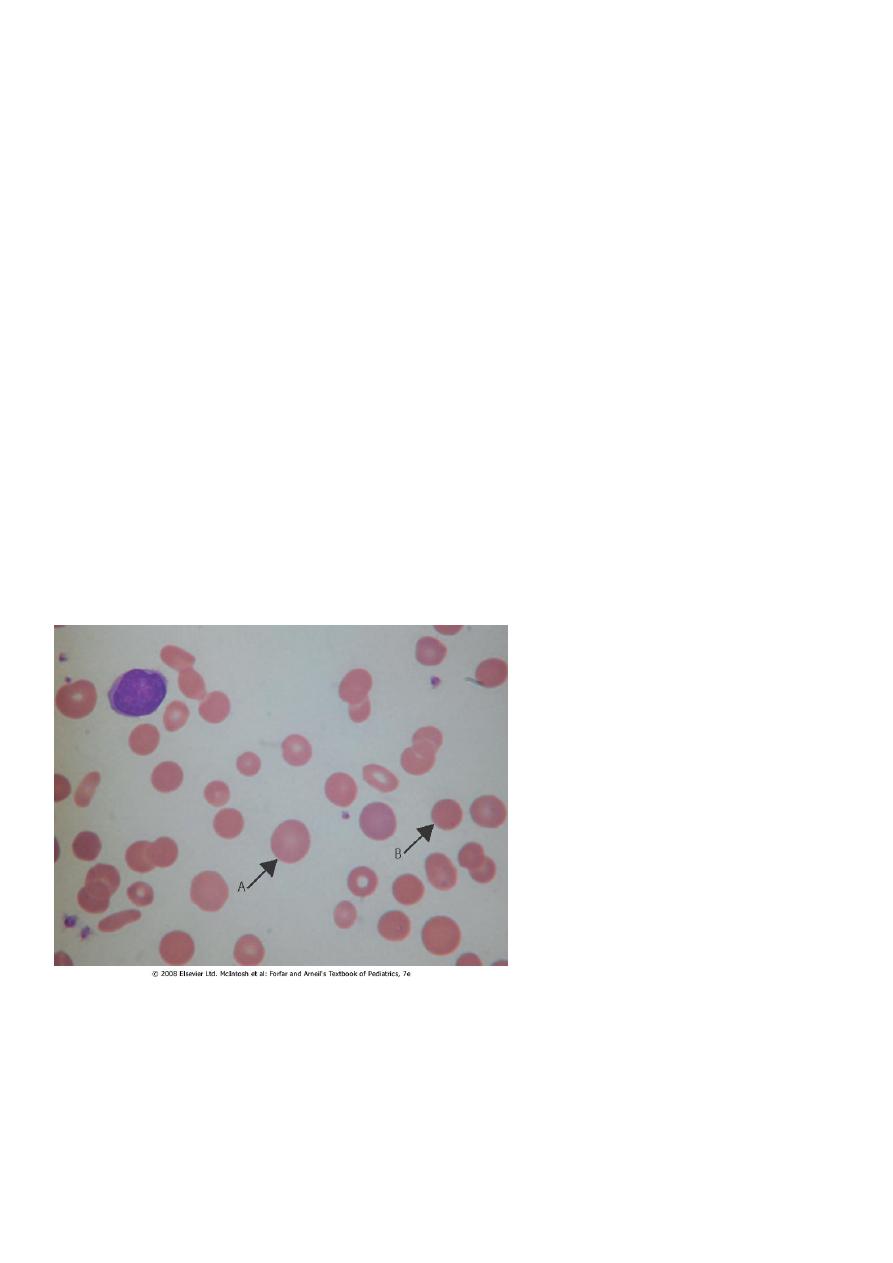
The characteristic finding is increased numbers of spherocytes in the peripheral blood. It is
inherited as A.D in 75% of cases, other cases inheritant as A.R or new mutation.
Pathology
In AD type the defect may be in either beta spectrin,ankyrin or protein 3. In AR form the
defect is in either -protein or protein 4-2.
This defect affect the RBC skeleton leading to budding of RBC memb, This bud is removed
rapidly by the RES leading to loss of RBC surface area.The stretching ability of the RBC is
very limited which lead to RBC rupture.
The spherocyte have no easy transit through the splenic cord because of its shape this will
affect glucose metabolism and decrease pH of RBC.
A, polychromatic cell; B,
microspherocyte

4
Clinical pictures
1. May be asymptomatic with mild anemia & discovered accidentally.
2. The severe form may be started at early infancy with neonatal jaundice, severe anemia ,
splenomegaly & chronic blood transfusion.
3. Clinical pictures of complication, which are:
A- Bone marrow aplasia after Parvo virus B19 infection: This v. infect erythroid cells in the
BM & with arrest in development. The aplasia last for 10-14 days during which time the Hb
drops by 50% with reticulocytopenia.
B- Hyper hemolysis after some viral infection lead to increasing anemia, reticulocytosis
&jaundice.
C- Delayed growth and sexual developments.
D- Pigmented gallstones.
Laboratory diagnosis
1- CBC: A- anemia.
B- high MCHC>36.
C- high retic. count.
D- normal MCH & MCV.
E- spherocytes in peripheral blood.
2- Incubated osmotic fragility test is elevated.
3- High indirect bilirubin.
4- Decrease habtoglobin.
5- Membrane protein analysis for difficult cases
Management
1- In mild anemia with normal growth and development,we only observe the child and limit
the intervention to folic acid tablet 1 mg/day.
2- Splenectomy for any child with chronic anemia or growth failure but we should defer it
till the age of 5 yr to minimize the risk of sepsis & we should vaccinate the child by
pneumococcal , H. influenza & menengiococcal vaccines at least 2 weeks before
splenectomy & give the child prophylactic penicillin after splenectomy until at least 18 yr of
age.

5
In some reports,partial splenectomy appears to improve the anemia & maintain splenic
function.
3- Elective cholecystectomy for symptomatic gallstones.
Glucose-6-phosphate dehydrogenase(G6PD) deficiency
G6PD deficiency is the most common RBC metabolic disorder. It is usually transmitted in an
X-linked recessive fashion.
Defects: The two prototypic forms are:
The A- variant is found mainly in the black population and is associated with an isoenzyme
that deteriorates rapidly (it has a half-life of 13 days).
The Mediterranean variant is found mainly in individuals of Greek and Italian descent and
is associated with almost complete absence of enzyme activity, even in young cells, due to
extreme instability (it has a half-life of several hours).
Pathogenesis
G6PD-deficient cells do not generate an amount of reduced glutathione that is sufficient to
protect the RBCs from oxidant agents.
Exposed sulfhydryl groups of Hb are oxidized, predisposing the molecule to denaturation.
The heme and globin moieties dissociate, with the globin precipitating as Heinz bodies.
The damaged RBCs are then removed by the RES; severely damaged cells may lyse
intravascularly.
HA results from oxidative damage to the RBCs as consequence of the loss of the protective
effect of the enzyme G6PD.
The prevalence of G6PD def. is related to the prevalence of malaria as in Africa, it also high
in mediterranean area. The incidence of P falceparum parasite is lower in G6PD def.
patients.
The half-life of the enz in normal RBC is 60 days, the mature RBC cannot synthesize the enz.
The younger RBCs are relatively more resistant to hemolysis .
The deficient RBC hemolysed when exposed to exogenous factors. The particles of the
denaturated Hb, Heinz body, attach to the cell membrane causing irreversible damage and
lysis, most of lysis occur intravascularly causing hemoglobinemia & hemoglobinuria.
There may be an extravascular hemolysis which explain the splenomegaly in some cases.

6
Favism
is the classical cause of acute hemolysis in G6PD def. Fava bean contain the beta glycosides
vicine & convicine These substances may undergo auto-oxidation as part of their
metabolism, producing free O2 radicals.
Acute hemolysis that occurs upon exposure to Fava is characterized by:
1- Unpredictable (only 25%of adult at risk develop hemolysis).
2- Influence of dose and body weight.
3- Maturity of the bean.
4- Quality of bean (raw beans more than cooked, frozen or canned beans)
5- The activity of beta glucosidases in the bean & intestinal mucosa.
Drug induced hemolysis
Many drugs and chemicals have been associated with hemolysis in G6PD def patient,These
substances have the ability to stimulate the pentose phosphate PW in RBC which can lead
to oxidation of NADPH .
Infection-caused hemolysis
During the process of phagocytosis of bacteria there is a release of peroxides by the
phagocytosing granules, these peroxides lead to release of O radicals.
Clinical presentation
There are three primary clinical presentation:
1- Neonatal jaundice.
2- Acute hemolysis beyond the neonatal period.
3- Chronic hemolysis (congenital non-spherocytic HA).
1- Neonatal jaundice:
Acute hemolysis is characterized by onset of jaundice on the first few days of life that is out
of the proportion to the degree of anemia. All infant with G6PD def develop NJ.
NJ in G6PD def neonate may be an exaggerated physiological j or may be due to acute
hemolysis caused by inciting agent like inf., drugs, naphthalene ball used in stored cloth
diapers in developing country.

7
2-Acute hemolysis:
Most pt with G6PD def. are asymptomatic until exposed to inciting agent at which time
they may develop hemolysis. The onset of hemolysis is usually within 24-48 hr of exposure.
The initial manifestation may include abdominal pain, vomiting or diarrhea, tea colored
urine (hemoglobinuria),Jaundice, pallor with symptoms of anemia.
Examination usually reveals: anemia , jaundice splenomegaly and hepatomegaly & in severe
case symptom of heart failure.
Lab. finding in acute hemolysis include:
1- Normochromic normocytic anemia with anisopoikilocytosis, with few spherocyte .
2- Reticulocytosis.
3- Presence of bite cells (RBC bitten by macrophages).
4- Presence of hemigoast cells(RBC with uneven Hb distribution).
5- By using supravital stain we find inclusion bodies called Heinz body(denatured Hb).
6- Low serum haptoglobin.
7- High uncong. bilirubin.
8- Hemoglobinuria .
9- hemoglobinemia.
Diagnosis of G6PD def
1- Antenatal diagnosis can be made by performing G6PD assay on amniotic fl cells or from
chorionic villi biopsy and DNA assay.
2- Screening test: semi quantitative test, this test should not be done during acute
hemolysis because it lead to normal value. The usual cut of deficiency value is <30% of
normal activity.
3- Quantitative assay: done several weeks after acute hemolysis when reticulocyte return
to normal.
4- G6PD electrophoresis.

8
Differential diagnosis:
1- AIHA.
2- Malaria induced hemolysis.
3- Hepatitis
Management of acute severe hemolysis
1- Removal of the inciting agent.
2- Brisk hydration to ensure adequate urine output.
3- blood transfusion.
4- Folic acid tablet 1mg/d for one month.
3-Chronic hemolysis
Small minority of pt develop chronic H. Clinically all pts are male, presented with:
1-Neonatal j with anemia.
2-Jaundice and anemia.
3-splenomegaly.
4-Gallstone.
5-Acute hemolysis after exposure to oxidative agent.
Lab. Finding:
1- CBC: anemia, reticulocytosis & polychromasia.
2- Low haptoglobin.
3- Hyberbilirubinemia.
Management
1-Folic acid administration.
2-Blood transfusion if indicated.

9
Drugs induce hemolysis in G6PD:
1. ANALGESICS/ANTIPYRETICS acetophenetidin , amidopyrine ,aspirin ,phenacetin,
probenecid.
2. ANTIMALARIALS chloroquine, hydroxychloroquine mepacrine, pamaquine, pentaquine,
primaquine, quinine, quinocide
3. SULFONAMIDES/SULFONES dapsone, sulfacetamide, sulfamethoxypyrimidine,
sulfanilamide ,sulfapyridine sulfasalazine ,sulfisoxazole .
4. ANTIBACTERIAL chloramphenicol, cotrimoxazole, furazolidone, nalidixic acid,
nitrofurantoin nitrofurazone , para-aminosalicylic acid.
5. MISCELLANEOUS alpha-methyldopa, ascorbic acid dimercaprol (BAL), hydralazine ,
methylene blue ,nalidixic acid ,naphthalene, vitamin K (water soluble)