Fifth stage
PediatricLec. 5
د. انور
12/4/2017
Hb S disorders(Sickle cell disorders)
Defect and pathogenesis
The molecular defect is the result of an abnormal autosomal gene that substitutes valine for glutamic acid in the 6th position of the β-globin chain.
Under conditions of hypoxia, the Hb aggregates into insoluble long polymers that align themselves into rigid paracrystalline gels , which distort the RBC into a sickle shape.
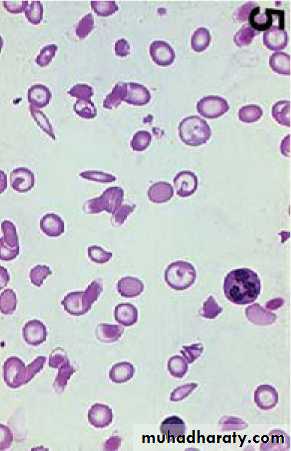
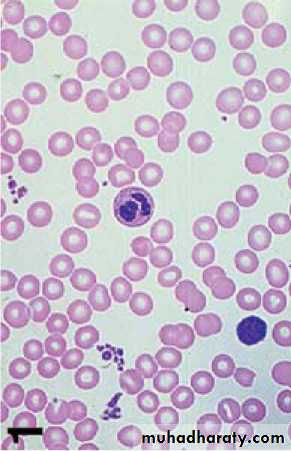
The clinical consequences of the solubility anomaly are:
1- Shortened red blood cell survival (hemolytic anemia)2- Microvascular obstruction, which leads to tissue ischemia and infarction
There are two types of Hb S disease:
1- Heterozygous state (sickle cell trait )
2- Homozygous state (sickle cell anemia)
Heterozygous state (sickle cell trait)
Both Hb A and Hb S exist in individuals who have sickle cell trait; there is more HbA than HbS.Clinical features:
Sickle cell trait is usually asymptomatic, unless the affected individual is subjected to hypoxemic stress. Otherwise, abnormalities may be limited to:1- failure to concentrate urine.
2- painless hematuria, or both.
Diagnosis:
Sickle cell trait may be diagnosed by Hb electrophoresis or solubility tests (e.g slide test).It is important to detect the trait for purposes of genetic counseling.
Therapy:
No specific treatment is required; however, precautions to avoid hypoxemia associated with severe pneumonia, unpressurized flying, exercise at high altitudes, and G/A are in order. Tourniquet surgery and deep hypothermia should be avoidedHomozygous state (sickle cell anemia)
Clinical pictures:
1- In the asymptomatic period, the high levels of Hb F during fetal life and the first few months of postnatal life protect the patient.
2- Dactylitis : The earliest clinical manifestation may occur at 4 to 6 ms of age, when symmetric, painful swelling of the dorsal surfaces of the hands and feet develops. This is caused by avascular necrosis of the bone marrow of the metacarpal and metatarsal bones.
During this same period, progressive anemia with jaundice and splenomegaly begins to develop.
3- Splenic sequestration crises: The spleen may suddenly become engorged with RBCs, trapping a significant portion of the blood volume.
The child may presented with sudden severe pallor , severe abdominal pain and huge splenomegaly
If not corrected rapidly, this can lead to hypovolemic shock and death.
4- Overwhelming infections:
Pts are very susceptible to overwhelming infection, particularly with encapsulated bacteria such as pneumococci and Haemophilus influenzae; Salmonella septicemia and osteomyelitis are also seen with increased frequency in pts who have SCA.
5- Aplastic crises can occur at any age when there is suppression of erythropoiesis in response to a viral infection such as parvovirus B19 .
6- Vaso-occlusive episodes can involve any tissue. Depending on the involved organ.
A vaso-occlusive episode can produce abdominal pain, bone pain, cerebrovascular accident (CVA), pulmonary infarction (acute chest syndrome) , hepatopathy, or hematuria. These episodes are often precipitated by infection, dehydration, chilling, vascular stasis, or acidosis.
Repeated vaso-occlusive episodes in the spleen lead to infarction and fibrosis of this organ; it gradually regresses in size and is usually no longer palpable after the age of 5 years.
7- Late manifestations.
By the time a pt reaches his late teens or early 20s, he is suffering the long-term consequences of chronic anemia, and tissue infarction.
Many succumb to progressive myocardial damage with congestive heart failure.
Other long-term complications include gallstones, leg ulcers, renal damage, and aseptic necrosis of the long bones.
Treatment of SCA
1- Infections. Because these patients suffer from functional asplenia, the same precautions to protect them from overwhelming Gram-positive sepsis must be taken as for the patient whose spleen has been surgically removed.However, after the age of 5, there is little evidence to suggest that routine penicillin prophylaxis is required.
2-Vaso-occlusive episodes.
Prevention by avoidance of dehydration, hypoxia, chilling, and acidosis.Treatment is as follows:
-Analgesics should be given for pain.
-When a vital organ (the brain, liver, or lung) is threatened, transfusion with packed RBCs may be necessary.
-After a documented CVA, the pt remains at high risk for recurrent CVAs for an indefinite period of time; such pts should be maintained on a chronic transfusion program designed to keep the Hb S level at <30%.
As is the case with chronic transfusion programs for pts who have thalassemias, iron overload may eventually necessitate chelation therapy
3- Use of agents that elevate Hb F levels.
Because Hb F levels correlate inversely with disease severity, efforts have been made to identify medications that might increase Hb F levels in sickle cell anemia pts.
Hydroxyurea has become the drug most commonly used for this purpose; early trials have indicated improvement in both laboratory and clinical parameters for children treated with this agent.
4- Severe aplastic crises should be treated by transfusion with packed RBCs .
5- HSC transplantation from healthy, HLA-matched sibling donors has proved to be potentially curative; however, this approach is limited by the paucity of HLA-matched siblings and the toxicity of the conditioning regimens.Antibody-mediated hemolytic anemias
General considerations1- Autoimmune hemolytic anemias (AIHA):
are the result of antibodies generated by an individual's immune system against his or her own RBCs.
2- Isoimmune hemolytic anemias:
result from antibodies produced by one individual against the RBCs of another individual of the same species. It can be seen in hemolytic disease of the newborn or hemolytic transfusion reactions (e.g., the transfusion of type A blood into an individual who has type B blood).
Autoimmune hemolytic anemias
Etiology :
AIHA may be idiopathic or the result of infectious agents, drugs, lymphoid neoplasms, or disorders of immune regulation (e.g., SLE, agammaglobulinemia)
Typical antibodies involved
1-Antibodies of the IgG class, for the most part, are warm reactive (i.e., they have maximal activity at 37°C). They are detected using the direct antiglobulin (Coombs) test.-These are incomplete antibodies in that they do not agglutinate RBCs, although they coat the surface.
-Hemolysis occurs extravascularly .
-IgG antibodies are associated clinically with autoimmune diseases, lymphomas, and viral infections. Occasionally, no underlying cause is demonstrable.
2-Antibodies of the IgM class are usually cold reactive (i.e., most have maximal activity at low temperatures).
-These are complete antibodies in that they agglutinate RBCs and activate the complement sequence through C9, causing lysis of RBCs.
-Hemolysis occurs intravascularly .
-IgM antibodies are associated clinically with mycoplasmal pneumonia, Epstein-Barr virus, and transfusion reactions.
3-Donath-Landsteiner antibody:
-It is of the IgG type, but it is exceptional in that it reacts best in the cold and can activate complement, causing hemolysis to occur intravascularly .
-Its clinical associations include syphilis and viral infections.
It may also be idiopathic.
Therapy:
depends on the cause, clinical condition of the pt, & expected duration of the illness. Because most cases of childhood AIHA are idiopathic or postinfectious and self-limited, supportive care and judicious use of transfusions and corticosteroids are the therapies most commonly used.Treatment modalities include:
Supportive care with bed rest and oxygen
Transfusion with packed RBCs
Corticosteroids
Splenectomy
Immunosuppressive agents