Autoimmune Connective Tissue DiseasesPart 2
Dr. Ali Abdul-Rahman YounisRHEUMATOLOGIST(FIBMS)
Systemic Sclerosis
Systemic sclerosis, or scleroderma, is a generalized disorder of connective tissue affecting the skin, internal organs and vasculature.It is characterised by sclerodactyly in combination with Raynaud’s, digital ischaemia and cardiac, lung, gut and renal disease.
It is subdivided into diffuse cutaneous systemic sclerosis (DCSS: 30% of cases) and limited cutaneous systemic sclerosis (LCSS: 70% of cases).
Many patients with LCSS have features that are phenotypically grouped into the ‘CREST’ syndrome (Calcinosis, Raynaud’s, Esophageal involvement, Sclerodactyly and Telangiectasia).
Systemic Sclerosis
The peak age of onset is in the fourth and fifth decades.Overall prevalence is 10–20 per 100 000.
4 : 1 female preponderance.
The prognosis in DCSS is poor, with a 5year survival of approximately 70%.
Features that associate with a poor prognosis include older age, diffuse skin disease, proteinuria, high ESR, a low TLCO (gas transfer factor for carbon monoxide) and pulmonary hypertension.
Pathophysiology
The cause of systemic sclerosis is poorly understood.There is evidence for a genetic component, and associations with alleles at the HLA locus have been found.
The disease occurs in all ethnic groups, and race may influence severity, since DCSS is significantly more common in black women than white.
Isolated cases have been reported in which a systemic sclerosis like disease has been triggered by exposure to silica dust, vinyl chloride, hypoxyresins and trichloroethylene.
There is clear evidence of immunological dysfunction: T lymphocytes infiltrate the skin and there is abnormal fibroblast activation, leading to increased production of extracellular matrix in the dermis, primarily type I collagen. This results in symmetrical thickening, tightening and induration of the skin (sclerodactyly).
Arterial and arteriolar narrowing occurs due to intimal proliferation and vessel wall inflammation. Endothelial injury causes release of vasoconstrictors and platelet activation, resulting in further ischaemia, which is thought to exacerbate the fibrotic process.
Clinical features
SKIN
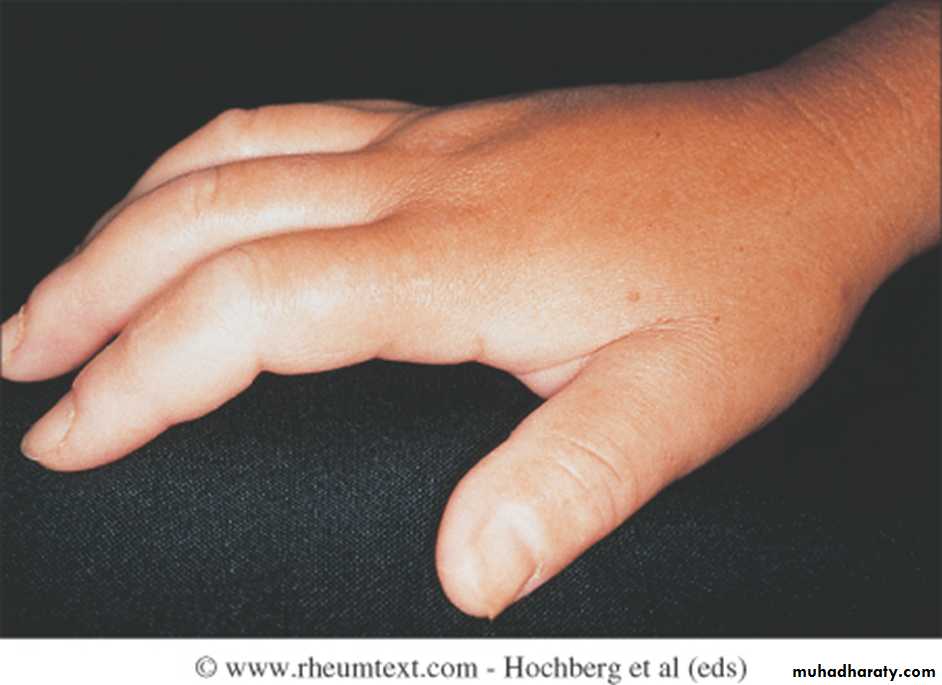
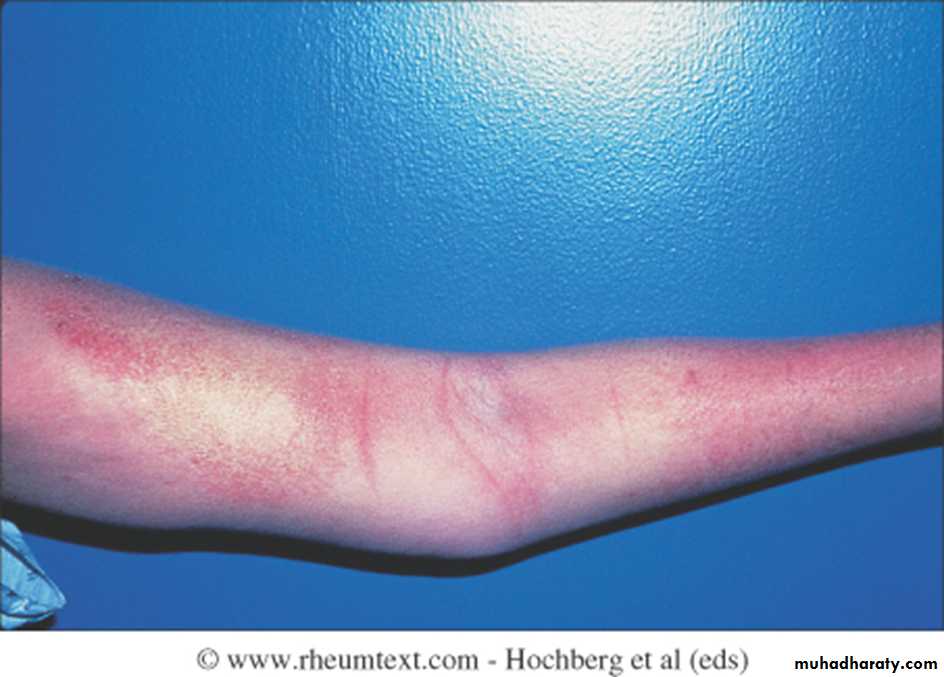
Initially, there is nonpitting oedema of fingers and flexor tendon sheaths. Subsequently, the skin becomes shiny and taut, and distal skin creases disappear.
This is accompanied by erythema and tortuous dilatation of capillary loops in the nailfold bed, readily visible with an ophthalmoscope or dissecting microscope (and oil placed on the skin).
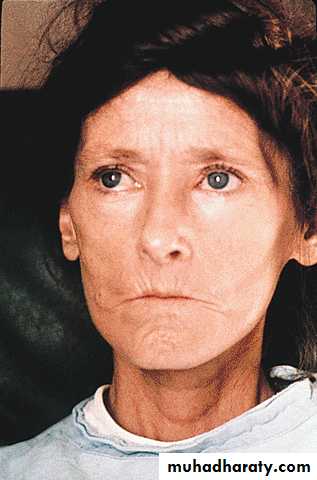
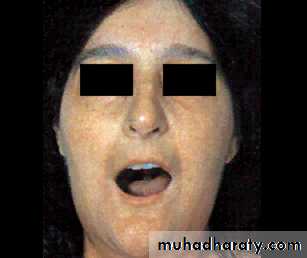
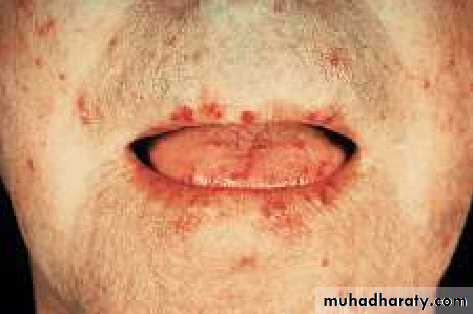
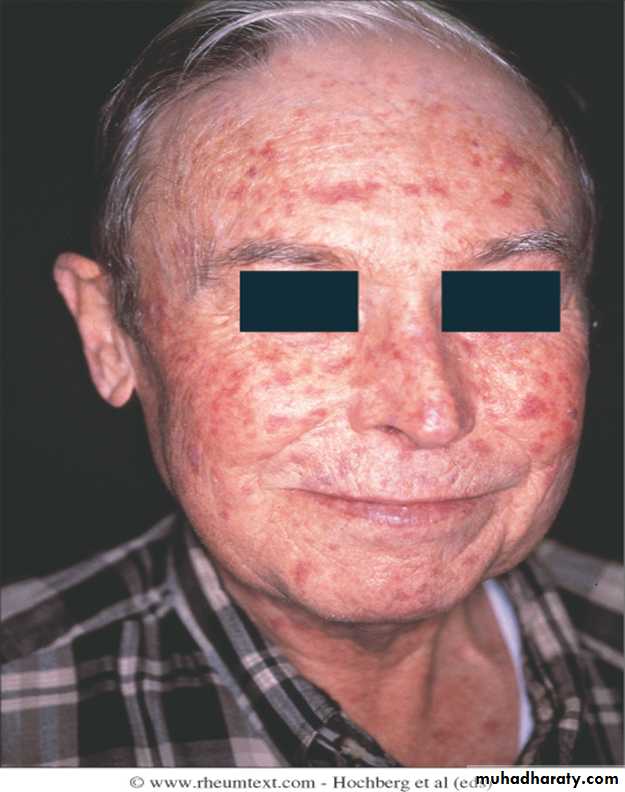
The face and neck are usually involved next, with thinning of the lips and radial furrowing. In some patients, skin thickening stops at this stage.
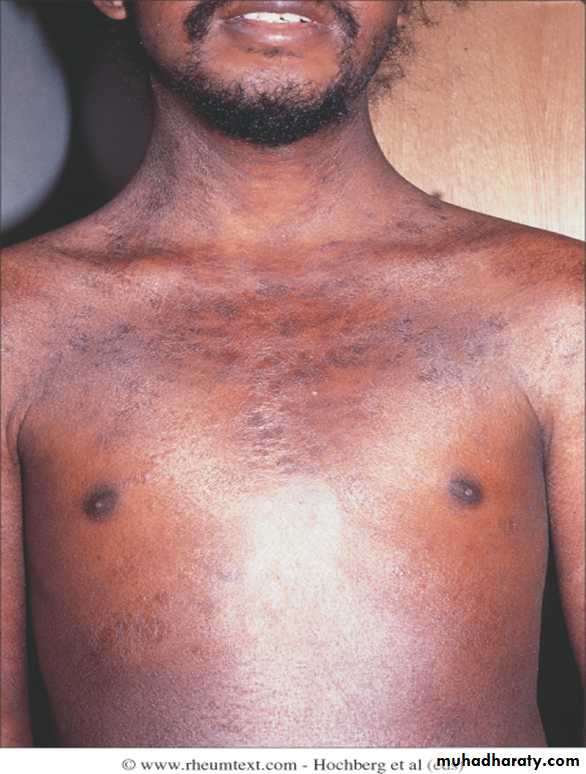
Skin involvement restricted to sites distal to the elbow or knee (apart from the face) is classified as ‘limited disease’ or CREST syndrome . Involvement proximal to the knee and elbow and on the trunk is classified as ‘diffuse disease’.
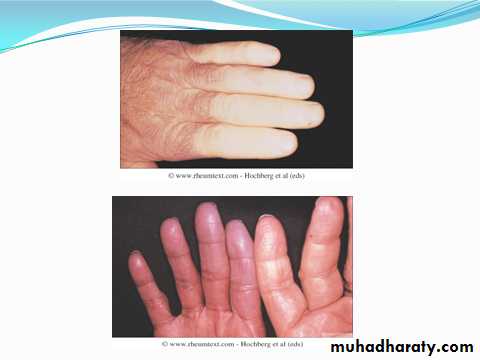
Raynaud’s phenomenon
This is a universal feature and can precede other features by many years.Involvement of small blood vessels in the extremities may cause critical tissue ischaemia, leading to skin ulceration over pressure areas, localized areas of infarction and pulp atrophy at the fingertips.
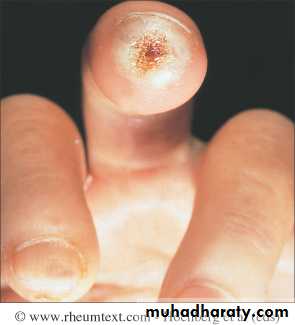
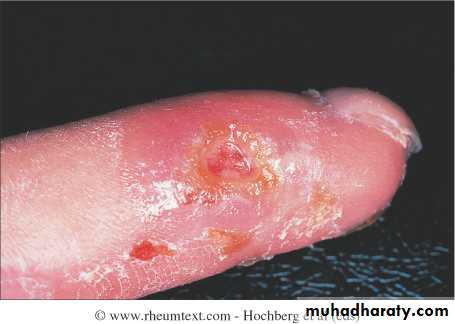
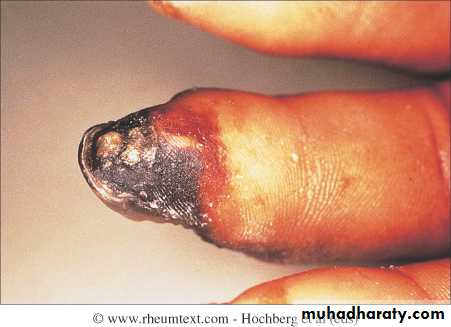
Musculoskeletal features
Arthralgia, morning stiffness and flexor tenosynovitis are common.Restricted hand function is due to skin rather than joint disease and erosive arthropathy is uncommon.
Muscle weakness and wasting can occur due to myositis.

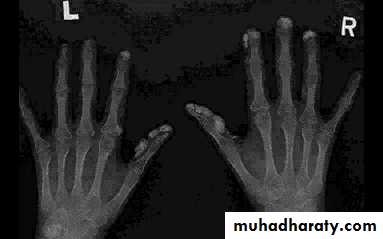
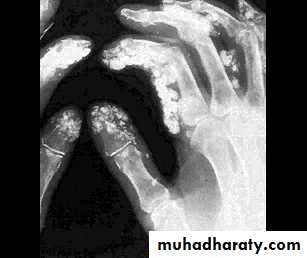
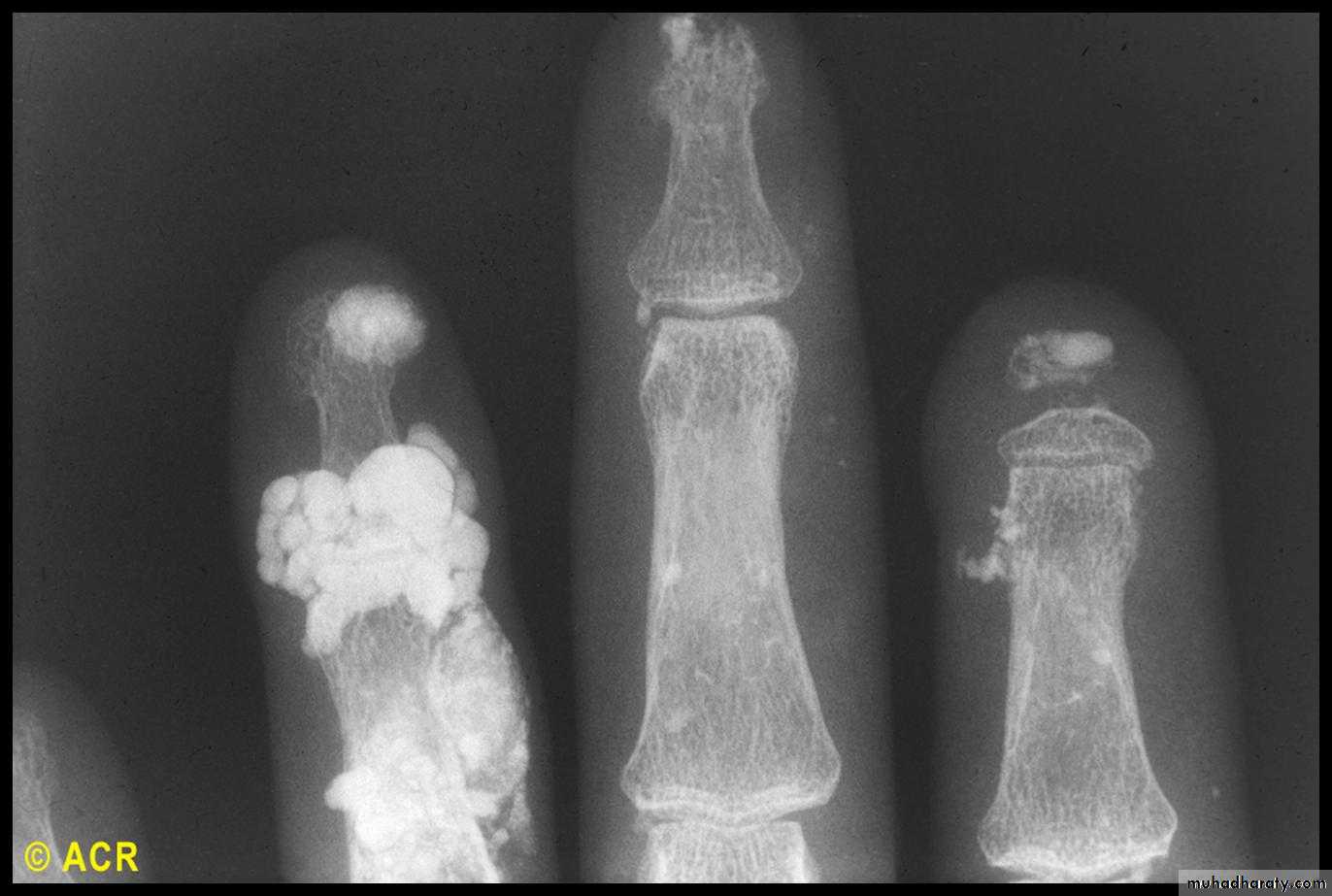
Gastrointestinal involvement
Smooth muscle atrophy and fibrosis in the lower two thirds of the oesophagus lead to reflux with erosive oesophagitis. Dysphagia and odynophagia may also occur.Involvement of the stomach causes early satiety and occasionally outlet obstruction. Recurrent occult upper gastrointestinal bleeding may indicate a ‘watermelon’ stomach (antral vascular ectasia), which occurs in up to 20% of patients.
Small intestine involvement may lead to malabsorption due to bacterial overgrowth and intermittent bloating, pain or constipation.
Dilatation of large or small bowel due to autonomic neuropathy may cause pseudoobstruction with nausea, vomiting, abdominal discomfort and distension, often worse after food.
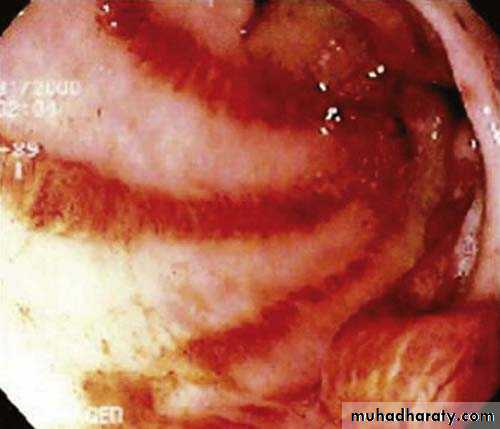
Pulmonary involvement
This is a major cause of morbidity and mortality.Pulmonary hypertension complicates longstanding disease and is six times more prevalent in LCSS than in DCSS.
It presents with rapidly progressive dyspnoea (more rapid than interstitial lung disease), right heart failure and angina, often in association with severe digital ischaemia.
Fibrosing alveolitis mainly affects patients with DCSS who have topoisomerase 1 antibodies.
Others: aspiration pneumonia, bronchiectesis, hidebound chest, alveolar and bronchial carcinoma.
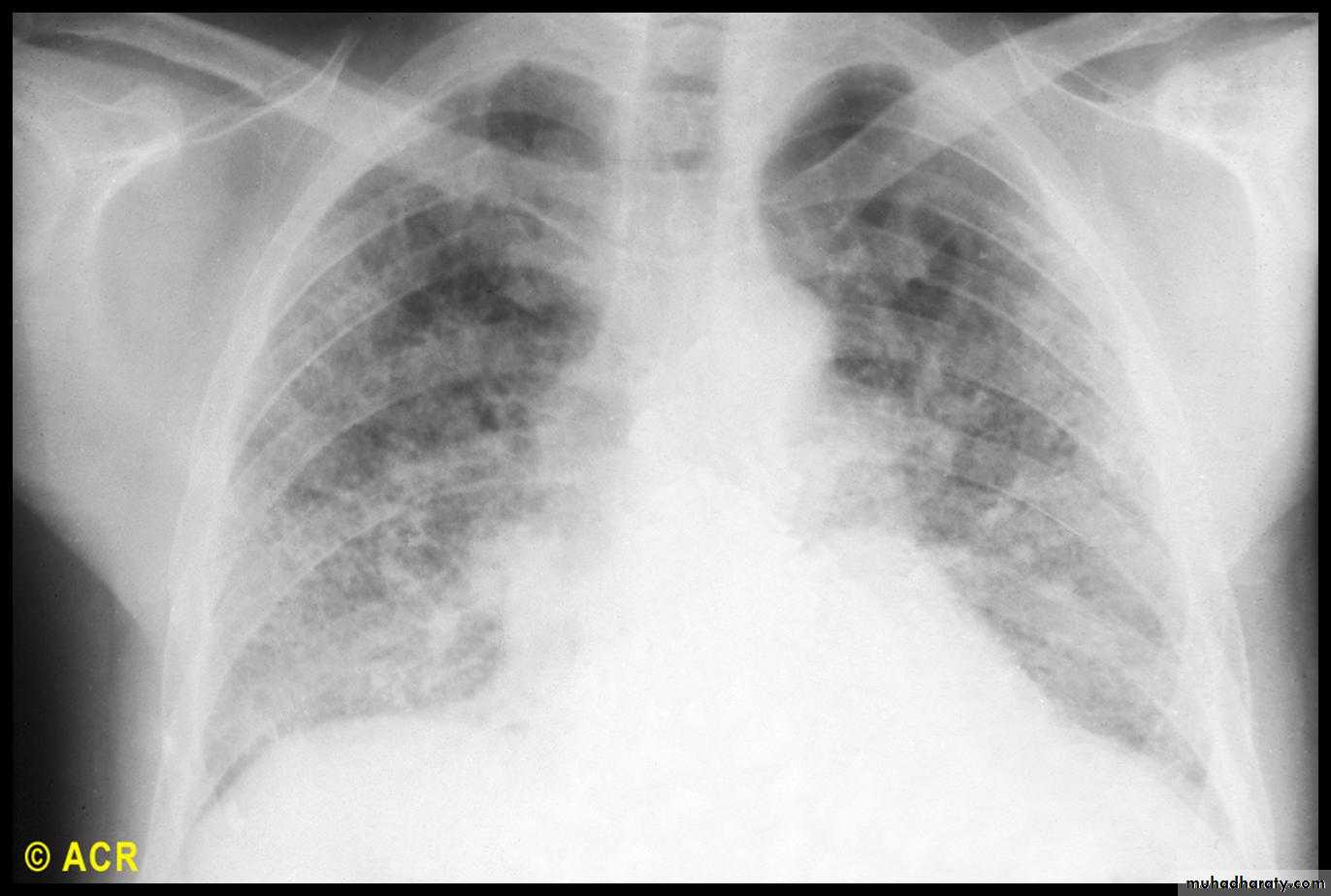
Cardiac features
Pericarditis (with or without effusion)Heart failure
Arrhythmias
Cardiomyopathy
Systemic hypertension or pulmonary hypertension
Renal involvement
One of the main causes of death is hypertensive renal crisis, characterized by rapidly developing accelerated phase hypertension and renal failure.
Hypertensive renal crisis is much more likely to occur in DCSS than in LCSS, and in patients with topoisomerase 1 antibodies and RNP antibodies.
Investigations
Scleroderma is primarily a clinical diagnosis but various laboratory abnormalities are characteristic.The ESR is usually elevated and raised levels of IgG are common, but CRP values tend to be normal unless there is severe organ involvement or coexisting infection.
ANA is positive in about 70%, and approximately 30% of patients with DCSS have antibodies to topoisomerase 1 (Scl70).
About 60% of patients with CREST syndrome have anti
centromere antibodies
Investigations
Chest X-ray, transthoracic echocardiography and lung function tests are recommended to assess for interstitial lung disease and pulmonary hypertension.High-resolution lung CT is recommended if interstitial lung disease suspected .If pulmonary hypertension is suspected, right heart catheter measurements should be arranged at a specialist cardiac centre.
A barium swallow can assess oesophageal involvement. A hydrogen breath test can indicate bacterial overgrowth.
Management
No treatments are available that halt or reverse the fibrotic changes that underlie the disease.The focus of management, therefore, is to ameliorate the effects of the disease on target organs.
Raynaud’s should be treated by avoidance of cold exposure and use of mittens (heated mittens are available), supplemented if necessary with calcium antagonists.
Intermittent infusions of prostacyclin may benefit severe digital ischaemia.
The endothelin 1 antagonist bosentan can be of value in promoting healing of digital ulcers.
If these become infected, antibiotics may be required, but as these penetrate tissues poorly in scleroderma, they need to be given at higher doses for a longer duration than usual.
Management
Oesophageal reflux should be treated with proton pump inhibitors and antireflux agents. Antibiotics may be required for bacterial overgrowth syndromes, and metoclopramide or domperidone may help patients with symptoms of pseudoobstruction.
Hypertension should be treated aggressively with ACE inhibitors, even if renal impairment is present.
Joint involvement may be treated with analgesics and/or NSAID. If synovitis is present, immunosuppressants such as methotrexate can also be of value.
Management
Pulmonary hypertension may be treated with bosentan. In selected patients, heart–lung transplantation may be considered.Corticosteroids and cytotoxic drugs are indicated in patients who have coexisting myositis or fibrosing alveolitis.
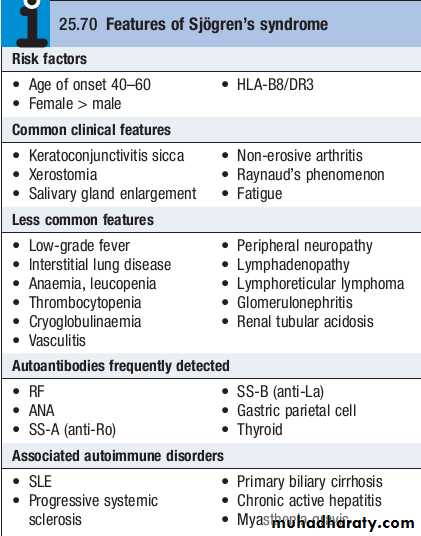
Sjögren’s syndrome
This is an autoimmune disorder of unknown cause, characterized by lymphocytic infiltration of salivary and lachrymal glands, leading to glandular fibrosis and exocrine failure.The typical age of onset is between 40 and 50, with a 9 : 1 female preponderance.
The disease may occur in isolation (primary Sjögren’s syndrome) or in patients with other autoimmune diseases (secondary Sjögren’s syndrome).
Clinical features
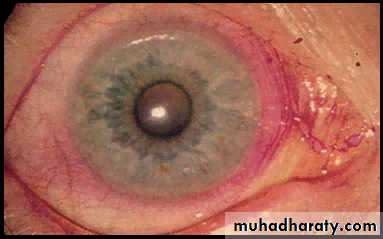
The eye symptoms, termed keratoconjunctivitis sicca, are due to a lack of lubricating tears .Conjunctivitis and blepharitis are frequent, and may lead to filamentary keratitis due to tenacious mucous filaments binding to the cornea and conjunctiva.
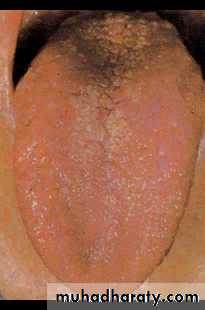
Oral involvement manifests as a dry mouth and typically the patient needs to sip water to swallow food. There is a high incidence of dental caries.
The disease is associated with a 40fold increased lifetime risk of lymphoma.
Investigations
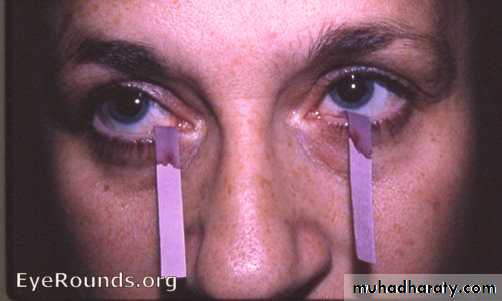
The diagnosis can be established by the Schirmer tear test, which measures tear flow over 5 minutes using absorbent paper strips placed on the lower eyelid; a normal result is more than 6 mm of wetting.Staining with rose Bengal may show punctate epithelial abnormalities over the area not covered by the open eyelid.
If the diagnosis remains in doubt, it can be confirmed by lip biopsy, which shows focal lymphocytic infiltrate of the minor salivary glands.
Most patients have an elevated ESR and hypergammaglobulinaemia, and one or more autoantibodies, including ANA and RF. AntiRo and antiLa antibodies are commonly present.
Management
No treatments that have disease modifying effects have yet been identified and management is symptomatic.Lachrymal substitutes, such as hypromellose, should be used during the day in combination with more viscous lubricating ointment at night.
Artificial saliva and oral gels can be tried for xerostomia but are often not effective.
Adequate postprandial oral hygiene and prompt treatment of oral candidiasis are essential.
Management
A trial of systemic pilocarpine (5–30 mg daily in divided doses) is worthwhile in early disease to amplify glandular function.
Hydroxychloroquine (200 mg twice daily) is often used to address skin and musculoskeletal features and may help fatigue.
Extraglandular and musculoskeletal manifestations may respond to steroids, and if so, immunosuppressive drugs can be added for their steroid sparing effect.
Immunosuppression does not improve sicca symptoms but is essential for progressive interstitial lung disease (e.g. glucocorticoids and cyclophosphamide) and for interstitial nephritis .
If lymphadenopathy or salivary gland enlargement develops, biopsy should be performed to exclude malignancy.