Sexual differentiation
Differentiation of the fertilized embryo into a male or female fetus is controlled by the sex chromosomes.All normal fetuses have an undifferentiated gonad which has the potential to become either a testis or an ovary. In addition, all fetuses have both Mullerian and Wolffian ducts and the potential to develop male or female internal and external genitalia.
The chromosomal complement of the zygote determines whether the indifferent gonad becomes a testis or an ovary. The first step in this pathway is dependent on the SRY gene (sex determining region of the Y chromosome). This gene, helped by other testes-determining genes, causes the gonad to begin development into a testis.
The Sertoli cells produce antiMullerian hormone (AMH) and the Leydig cells produce testosterone. Anti-Mullerian hormone suppresses development of the Mullerian ducts. Testosterone stimulates the Wollfian ducts to develop into the vas deferens, epididymis and seminal vesicles. In addition, in the external genital skin, testosterone is converted by an enzyme called 5-alpha-reductase into dihydrotestosterone (DHT). This acts to virilize the external genitalia.
The genital tubercle becomes the penis and the labioscrotal folds fuse to form the scrotum..
Where the gonad becomes an ovary, the absence of AMH allows the Mullerian structures to develop. bilateral Fallopian tubes, and the uterus, cervix and upper vagina. The rudimentary distal vagina fuses with the posterior urethra at week 7 to form the urogenital sinus.
Granulosa cells convert androgens ( coming from theca cells) to estrogen by aromatase during follicular phase Of the menstrual cycle.
Estrogen production :
Estrogen can be produced by fat tissue , liver, adrenal glands and the ovaries.Estrogen production from the ovaries begin with the theca interna cels , which convert cholesterol into hormone called androstiendione . this hormone is then exported to the granulose cells, these cells convert the androsteindione into estradiol
Ovary secret estrogen responsible for sexual characteristic and development of internal and external organs.
It is the main sex hormones in women and it is essential to the menstrual cycle.
Secondary sex characteristics which are the defining differences between men and women that do not relate to the reproductive system , are determined in part by estrogen.
In women these characteristics include breasts , widened pelvis and increased amounts of body fat in the buttock , thigh, and in hip region.
This hormone also contribute to the fact that women have less facial hair and smoother skin than men.
It is also an essential part of a woman′ s reproductive process.
(Estrogen regulate the menstrual cycle and prepare the uterus for pregnancy by enriching and thickening the endometrium).
During a woman′s reproductive life which starts with the onset of menstruation and continues until the menopause , the main type of estrogen produced is estradiol.
The luteinizing hormone and the follicle stimulating hormone help to control how the body produce estrogen in women who ovulate.
There are three distinct compounds that make up this hormone group: estrone , estradiol, and estriol.
Disorders of sex development
Disorders of sex development are conditions where the sequence of events described above does not happen. These may be diagnosed at birth with ambiguous or abnormal genitalia, but may also be seen at puberty in girls who present with primary amenorrhoea or increasing virilization.
There has been a recent change in the terminology used to refer to these conditions. Older terms, such as ‘hermaphrodite’ and ‘intersex’, are confusing to both the clinician and patients, and in addition can be hurtful.
New Terminology for Disorders of Sex Development (DSD).
Previous Proposed Intersex Disorders of Sex Development Male pseudohermaphrodite 46XY, DSD Undervirilization of xy male Undermasculinization of xy male
Female pseudohermaphrodite 46 XX, DSD Overvirilization of an XX female Masculinization of an XX
female True hermaphrodite: Ovotesticular DSD
Chromosomal abnormalities
Turner syndrome
Turner syndrome which results from a complete or partial absence on one X chromosome (45XO). Turner syndrome is the most common chromosomal anomaly in females, occurring in 1 in 2500 live female births. Although there can be variation among affected women, most have typical clinical features including short stature, webbing of the neck and a wide carrying angle. Associated medical conditions include coarctation of the aorta, inflammatory bowel disease, sensorineural and conduction deafness, renal anomalies and endocrine dysfunction, such as autoimmune thyroid disease. In this condition, the ovary does not complete its normal development and only the stroma is present at birth. The gonads are called ‘streak gonads’ and do not function to produce oestrogen or oocytes.
Diagnosis is usually made at birth or in early childhood from the clinical appearance of the baby or due to short stature during childhood. However, in about 10 per cent of women, the diagnosis is not made until adolescence with delayed puberty. The ovaries do not produce oestrogen, so the normal physical changes of puberty cannot happen.
In childhood, treatment is focused on growth, but in adolescence it focuses on induction of puberty by hormone replacement therapy. Pregnancy is possible, but ovum donation is usually required. Psychological input and support is important.
XY gonadal dysgenesis
In this situation, the gonads do not develop into a testis, despite the presence of an XY karyotype. In about 10 per cent of cases, this is due to an absent SRY gene, but in most cases the cause is unknown. In complete gonadal dysgenesis (Swyer syndrome), the gonad remains as a streak gonad and does not produce any hormones. In the absence of AMH, the Mullerian structures do not regress and the uterus, vaginal and Fallopian tubes develop normally. The absence of testosterone mean the fetus does not virilize.The baby is phenotypically female, although has an XY chromosome. The gonads do not function and presentation is usually at adolescence with failure to go into spontaneous puberty. The dysgenetic gonad has a high malignancy risk and should be removed when the diagnosis is made. This is usually performed laparoscopically. Puberty must be induced with oestrogen.
Mixed gonadal dysgenesis is a more complex condition. The karyotype may be 46 XX, butmosaicism, e.g. XX/XY, is present in up to 20 percent.
In this situation, both functioning ovarian and testicular tissue can be present and if so this condition is known as ovotesticular DSD.
The anatomical findings vary depending on the function of the gonads.
For example, if the testes is functional, the the baby will virilize and have ambiguous or normal male genitalia.
The Mullerian structures are usually absent on the side of the functioning testes, but a unicorcuate uterus may be present if there is an ovary or streak gonad.
46XY DSD
Complete androgen insensitivity syndrome (CAIS) occurs in individuals where virilization of the external genitalia does not occur due to a partial or complete inability of the androgen receptor to respond to androgen stimulation. In the fetus with CAIS, testes form normally due to the action of the SRY gene. At the appropriate time, these testes secrete AMH leading to the regression of the Mullerian ducts. Hence, CAIS women do not have a uterus. Testosterone is also produced at the appropriate time, however, due to the inability of the androgen receptor to respond, the external genitalia do not virilize and instead undergo female development.
A female fetus is born with normal female external genitalia, an absent uterus and testes found at some point in their line of descent through the abdomen from the pelvis to the inguinal canal.
During puberty, breast development will be normal as a result of conversion of testosterone to estradiol, however, the effects of androgens are not seen, so pubic and axillary hair growth will be minimal.
Presentation is usually at puberty with primary amenorrhoea, although if the testes are in the inguinal canal they can cause a hernia in a younger girl. Once the diagnosis is made, initially management is psychological with full disclosure of the XY karyotype and the information that the patient will be infertile.
Gonadectomy is recommended because of the small long-term risk of testicular malignancy, although this can be deferred until after puberty. Once the gonads are removed, long-term hormone replacement therapy will be required.
The vagina is usually shortened and treatment will be required to create a vagina suitable for penetrative intercourse. Vaginal dilation is the most effective method of improving vaginal length . Surgical vaginal reconstruction operations are reserved for those women that have failed a dilation treatment programme
5-Alpha-reductase deficiency:
In this condition, the fetus has an XY karyotype and a normal functioning testes which produce both testosterone and AMH. However, the fetus is unable to convert testosterone to dihydrotestosterone in the peripheral tissues and so cannot virilize normally.Presentation is usually with ambiguous genitalia at birth, but can also be with increasing virilization at puberty of a female child due to the large increase in circulating testosterone with the onset of puberty.
In the Western world, the child is usually assigned to a female sex of rearing, but there have been descriptions of a few communities where transition from a female to male gender at puberty is accepted.
Congenital adrenal hyperplasia
This condition leads to virilization of a female fetus. It is due to an enzyme deficiency in the corticosteroid production pathway (production of cortisol from cholesterol ) in the adrenal gland with over 90 per cent being a deficiency in 21-hydroxylase, which converts progesterone to deoxycorticosterone, and 17-hydroxyprogesterone (17-OHP) to deoxycortisol.
The reduced levels of cortisol being produced lead to feedback mechanism on the hypothalamus lead to elevation of ACTH , resulting in hyperplasia of the adrenal glands and increased levels of progesterone production. This leads to an excess of androgen precursors and then to elevated testosterone production. Raised androgen levels in a female fetus will lead to virilization of the external genitalia.
The clitoris is enlarged and the labia are fused and scrotal in appearance. The upper vagina joins the urethra and opens as one common channel onto the perineum.
Signs and symptoms of classical congenital adrenal hyperplasia in infants include :
Ambiguous genitalia in girlsEnlarged penis in boys
Poor weight gain
Weight loss
Dehydration
Vomiting
In addition, two thirds of children with 21-OH CAH will have a ‘salt-losing’ variety. This represents a life threatening situation(adrenal crisis), and those children who are salt-losing often become dangerously unwell with seriously low levels of sodium , dehydration , diarrhea, vomiting , low blood sugar levels and shock within a few days of birth.
Affected individuals require lifelong steroid replacement, such as hydrocortisone – along with fludrocortisone for salt losers. Once the infant is well and stabilized on their steroid regime, surgical treatment of the genitalia is considered.
Surgery certainly leaves scarring and may reduce sexual sensitivity, but the alternative of leaving the genitalia virilized throughout childhood can be difficult for parents to consider.
At present, cases are managed individually by a multidisciplinary team involving surgeons, endocrinologists and psychologists.
Ambiguous genitalia in neonate
Be aware of associated metabolic problems .
Palpable gonads imply the presence of testicular tissue
Be very careful in your choice of words when talking to parents
The situation should be treated as a medical emergency, with pediatric endocrine advice being sought immediately
Investigations
Blood should be sent toChromosomal analysis
Gonadotrophins (LH,FSH)
Testosterone
Serum electrolyteand glucose
Serum 17 hydroxyprogestrone(17OHP) levels after day 3 of life
Pelvic and abdominal ultrasound : to determine pelvic structures and the presence or absence of gonads
Differential diagnosis:
Gonads palpable 46 XYGonadal dysgenesis
Partial androgen insensitivity
Biosynthetic defect in either testosterone or dihydrotestosterone production
Gonads impalpable 46 XX
Congenital adrenal hyperplasia
Gonadal dysgensis
exogenous androgen exposure
The decision as to the sex of rearing will be influenced by:
The baby′s karyotype
Gonadal status
Internal and external genital duct status
Potential for fertility
Cultural influences
Birth certificate wait until decision is made.
Do not complete the baby′s certificate until the sex of rearing has been decided.
If the wrong sex is entered on the form it will be extremely difficult to correct and require judicial intervention
Long term care
Families require long term medical and psychological support
Corrective surgery is usually undertaken within the first year of life but timing can be controversial
Infants with congenital adrenal hyperplasia have additional requirements for ongoing medical therapy
Disclosure to the patient about their diagnosis is usually undertaken in mid to late adolescence when they have the ability to understand complex issues such as chromosomes , hormones and posses some degree of emotional maturity
Mullerian anomalies
VAGINAL ABNORMALITIES
The significant abnormalities include:
Narrow introitus Hymen abnormality Septum Agenesis Associated abnormalities
NARROW INTROITUS
The existence is revealed after marriage. Dyspareunia may be the first complaint, or it may be detected during investigation of infertility. Treatment is effective by manual stretching under general anesthesia or by surgical enlargement .
HYMEN ABNORMALITY
Gross hymenal abnormality of significance is imperforate hymen. It is due to failure of disintegration of the central cells of the Müllerian eminence that projects into the urogenital sinus .
The existence is almost always unnoticed until the girl attains the age of 14–16 years. As the uterus is functioning normally, the menstrual blood is pent up inside the vagina behind the hymen (cryptomenorrhea). Depending upon the amount of blood so accumulated, it first distends the vagina (hematocolpos). The uterus is next involved and the cavity is dilated (hematometra). In the late and neglected cases, the tubes may also be distended after the fimbrial ends are closed by adhesions
Clinical features: The girl is aged about 14–16 years. The chief complaints are periodic lower abdominal pain, which may be continuous, primary amenorrhea and urinary symptoms, such as frequency, dysuria or even retention of urine. In fact, in significant cases the presenting feature may be the retention of urine. Abdominal examination reveals a suprapubic swelling, which may be uterine or full bladder. Prior catheterization reveals the true state. Vulval inspection reveals a tense bulging membrane of bluish coloration. Rectal examination reveals the bulged vagina. Ultrasonography can make the diagnosis of hematometra and hematocolpos.
Treatment Cruciate incision is made in the hymen. The quadrants of the hymen are partially excised not too close to the vaginal mucosa. Spontaneous escape of dark tarry colored blood is allowed Antibiotic should be given.
VAGINAL MALDEVELOPMENTS
Common variations of vaginal maldevelopments
♦ Agenesis of vagina ♦ Failure of vertical fusion ♦ Failure of lateral fusion
Pathology of Müllerian malformation: It may be due to failure of formation of the vaginal plate or due to its failure of canalization or cavitation. Transverse vaginal septa are due to faulty fusion or canalization of the urogenital sinus and the Müllerian ducts. About 45% occur in the upper vagina, 40% in mid vagina and 15% in the lower vagina. Septum located in the lower vagina is often complete and the signs and symptoms are similar to that of imperforate hymen. Ultrasonography is a useful investigation todetecthematometra, hematocolpos, and also urinary tract malformations. The principles of surgical treatment are the same. Septum in the upper vagina is often perforated. Incision of a complete (imperforate) septum becomes easy when the upper vagina is distended.
Longitudinal septum of the vagina may be present when the distal parts of the Müllerian ducts fail to fuse (fusion failure). It may be associated with double uterus and double cervix. It may be asymptomatic and needs no treatment. But it may cause dyspareunia or may obstruct delivery. In such circumstances, the septum is to be excised. Results of surgery are good in terms of achieving pregnancy.
PARTIAL AGENESIS OF UPPER VAGINA A segment of vagina may be atretic in the upper-third. It is often associated with hypoplasia or even absence of cervix. Uterus may be normal and functioning or malformed. Primary amenorrhoea (cryptomenorrhea) hematometra, hematocolpos, cyclic lower abdominal pain and presence of lower abdominal mass (as felt per abdomen or per rectum) point to the diagnosis. Conventional treatment is hysterectomy. Currently, abdominovaginal approach is made to establish communication between the uterovaginal canal above and the newly created vagina below. The result is, however, not always satisfactory though successful pregnancy and live birth have been reported. When hysterectomy is considered, ovaries should be conserved.
COMPLETE AGENESIS Complete agenesis of the vagina is almost always associated with absence of uterus. There is, however, presence of healthy gonads and fallopian tubes. The patient is phenotypically female, with normal female karyotype pattern. The entity is often associated with urinary tract (40%) and skeletal (12%) malformation. This is called Mayer-RokitanskyKüster-Hauser syndrome. The patient usually seeks advice for primary amenorrhea and dyspareunia.
Treatment of such patients needs repeated psychological counseling. Often they are depressed concerning their sexual and reproductive life. Treatment options are: (1) Nonsurgical, (2) Surgical.
1. Nonsurgical method: Repeated use of graduated vaginal dilators for a period of 6–12 months.
2. Surgical methods various procedures of vaginal reconstruction (vaginoplasty) are done.
UTERINE ANOMALIES
Uterine anomalies are often associated with vaginal maldevelopment.
Incidence of Müllerian abnormalities: It varies between 3 and 4%.. Failure of recanalization of the Müllerian ducts Agenesis of the upper vagina or of the cervix—This may lead to hematometra as the uterus is functioning
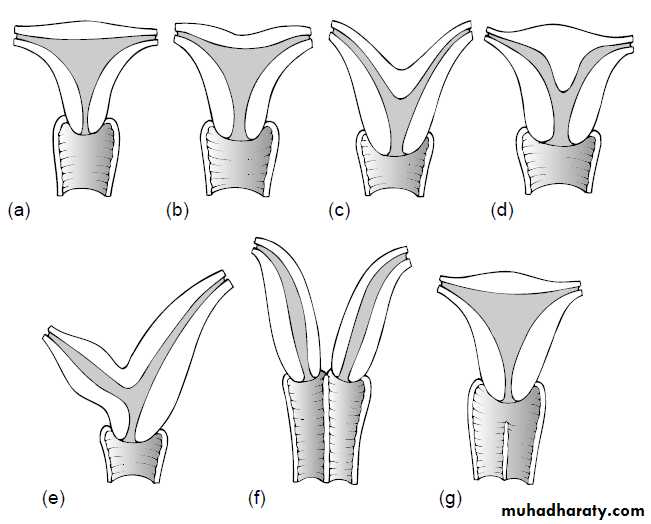
Types of fusion anomalies
-Arcuate (18%): The cornual parts of the uterus remains separated. The uterine fundus looks concave with heart-shaped cavity outline .
-Uterus didelphys (8%): There is complete lack of fusion of the Müllerian ducts with a double uterus, double cervix and a double vagina .
-Uterus bicornis (26%): There is varying degrees of fusion of the muscle walls of the two ducts. − Uterus bicornisbicollis: There are two uterine cavities with double cervix with or without vaginal septum. − Uterus bicornisunicollis: There are two uterine cavities with one cervix.
-Septate uterus (35%): The two Müllerian ducts are fused together but there is persistence of septum in between the two.
- Uterus unicornis (10%): Failure of development of one Müllerian duct
DES-related abnormality
Vagina: Adenosis, adenocarcinoma. Cervix: Cockscomb cervix, cervical collar. Uterus: Hypoplasia, T-shaped cavity, uterine adhesions. Fallopian tube: Cornual budding, abnormal fimbriae. Such cases are not seen now.
Clinical features
As previously mentioned, the condition may not produce any clinical manifestation.
Gynecological
Infertility and dyspareunia are often related in association with vaginal septum.
ii. Dysmenorrhea in bicornuate uterus or due to cryptomenorrhea (pent up menstrual blood in rudimentary horn).
iii. Menstrual disorders (menorrhagia, cryptomenorrhea) are seen..
Obstetrical
Midtrimester abortion which may be recurrent.ii. Rudimentary horn pregnancy may occur due to transperitoneal migration of sperm or ovum from the opposite side. Cornual pregnancy (ectopic) inevitably ends in rupture around 16th week.
iii. Cervical incompetence.
iv. Increased incidence of malpresentation— transverse lie in arcuate or subseptate, breech in bicornuate, unicornuate or complete septate uterus.
v. Preterm labor, IUGR, IUD.
Prolonged labor—due to incoordinate uterine action.
vii. Obstructed labor—obstruction by the nongravid horn of the bicornuate uterus or rudimentary horn.
viii. Retained placenta and postpartum hemorrhage where the placenta is implanted over the uterine septum.
Diagnosis:
Internal examination reveals septate vagina and two cervices. Passage of a sound can diagnose two separate cavities.
In fact in significant number of cases, the clinical diagnosis is made during uterine curettage, manual removal of placenta or cesarean section.
For exact diagnosis of the malformation, internal as well as external architecture of the uterus must be visualized. For this reason several investigations in different combinations are done, such as hysterography , hysteroscopy , laparoscopy , ultrasonography (vaginal probe) and magnetic resonance imaging (MRI). Ultrasonography and MRI are noninvasive procedures. Urological tract is also evaluated at the same time. The renal tract abnormality in association with Müllerian abnormality is about 40%. Skeletal system anomaly (12%) is also associated.
Treatment: Mere presence of any uterine malformation per se is not an indication of surgical intervention.
Reproductive outcome: Better obstetric outcome in septate uterus (86%), bicornuate uterus (50%) has been mentioned. Unicornuate uterus has very poor (40%) pregnancy outcome. No treatment is generally effective. Uterus didelphys has best possibility of successful pregnancy (64%). Other causes of infertility or recurrent fetal loss must be excluded. ƒ Rudimentary horn should be excised to reduce the risk of ectopic pregnancy (8%) ƒ Unification operation (bicornuate/septate uterus) is, therefore, indicated in otherwise unexplained cases with uterine malformation.
Abdominal metroplasty could be done either by excising the septum or by incising the septum . Success rate of abdominal metroplasty in terms of live birth is high (5–75%).
Hysteroscopicmetroplasty is more commonly done. Resection of the septum can be done either by a resectoscope or by laser . Advantages are: (a) High success rate (80–89%), (b) Short hospital stay, (c) Reduced postoperative morbidity (infection or adhesions), and (d) Subsequent chance of vaginal delivery is high compared to abdominal metroplasty where cesarean section is mandatory.