Lec: 4+5
د. خالد نافع28/10/2013
العدد (9)
HEMOGLOBINOPATHIES
Definition :Hemoglobinopathies are inherited disorders in which Mutation in or near the globin genes alter the structure of amino acid sequences(QUALITATIVE) Hb-S B6 glu val Hb-C B6 glu lys
or the rate of synthesis of a particular globin chain(QUANTITATIVE).
Defective haemoglobin
Sickle cell anaemia
It results from single base change in the DNA coding for the amino acid in the sixth position in the b-globin chain.
This leads to an amino acid change from glutamic acid to valine HbS will be formed instead of the normal Hb.
INTRODUCTION


Sickle Cell Anemia is a hereditary disease which is cause by a disorder in the blood, a mutation in the Hemoglobin Beta Gene which can be found in the chromosome 11. This disease causes the body to make abnormally shapes red blood cells. A normal red blood cell is shaped as a round donut while the abnormal red blood cell has a “ C “ form.
Hb S is insoluble and forms crystals when exposed to low oxygen tension.
Deoxygenated sickle Hb polymerizes into long fibrils.
The red cells sickle may block the different areas of the microcirculation or large vessels causing infarcts of various organs.
It is widespread in Africa.
ORIGIN
The origin sickle cell anemia is thought to be originated from Africa and India, and then started spreading as people move and mated.


Hemoglobin Beta Gene (HBB) also known as Beta Globin is a protein that resides in the red blood cells. The HBB is 146 amino acids long and its molecular weight is 15,867 Daltons. The molecules of the hemoglobin are responsible to carry oxygen through the body.
The HBB is found in part 15.5 of the chromosome 11.
RISK FACTORS
A risk factor is what increases the chance of getting a disease or condition.

The risk of having sickle cell anemia is dying of strokes and heart attacks dramatically increase.

Pathophysiological effects of sickled cells
1.Extravascular hemolysis . 2. Loss of splenic function. 3.Anaemia. Compromisation of the microcirculation.CLINICAL MANIFESTATION 1.complication from moderate to severe anaemia 2.slowed growth and development . 3.cardiac over load leads to CHF . 4.Bilirubin stones and cholecystitis .
Aplastic crisis.
Sickle cell crisis :

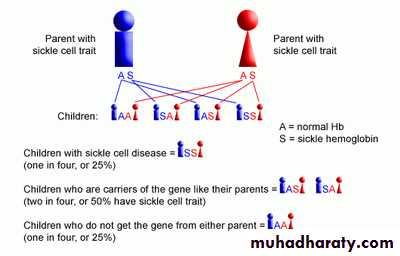
1.Splenic crisis (splenic sequestration syndrome, auto splenectomy) 2.Infections. 3.CNS and ophthalmic events (CVA, proliferative retinopathy). 4.Acute chest syndrome (chromic pulmonary hypertension lead to cor-pulmonale). 5.GIT : diffuse abdominal Pain. 6.Genitourinary symptoms: - Painless haematuria. - hyposthenuia. - priapism. - hypogonadism.

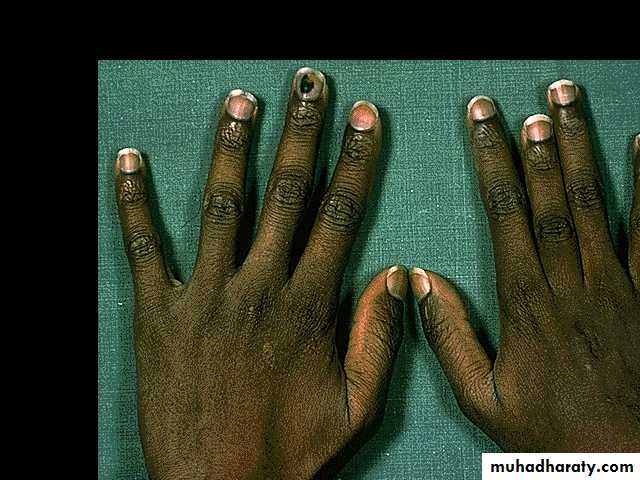
7.skeletal complication: - hand-foot syndrome. acute arthritis. aseptic necrosis. osteomyelitis . 8. Skin changes lead to chromic non- healing ulcer


Howell Jolly BodyErythroblastDiagnosis Peripheral blood smears : sickled cells, target cells, Howell-Jolly bodies, normoblast, red cell fragment, increase platelet and occasionally leukocytosis. screening test : sickling test Hemoglobin electrophoresis (Hb-S = 87% , Hb-F = 9.7%, Hb-A2 =3.3%)
Haemoglobin Electrophoresis (alkaline pH )Hb CHb SMoves in same position as Hb A2HbAAnodeHaemolysate appliedCathode
δδαααssααγγα Hb SHb A2Hb F Genotype αααα βsβs δδ γγHaemoglobins Produced :Diagnosis: Hb SS DiseaseLaboratory diagnosis of sickle cell anaemia made by presence of only Hb S, Hb A2, and Hb F on Hb electrophoresis with no Hb A, a positive sickling test and presence of sickle cells in blood film
Haemoglobin Electrophoresis:
Hb A 0 %Hb S 87.0 %
Hb F 9.7 %
Hb A2 3.3 %
Hypothesized protective mechanisms of sickle cell trait (HbAS) against malaria
P. falciparum ring-stage parasites did not grow in HbAS red blood cells under low oxygen tension.
specific intra-erythrocytic conditions of HbAS red blood cells, such as low intracellular potassium, high concentrations of haemoglobin or osmotic shrinkage of the red blood cell cause an inhospitable environment for parasites.
Recent data provide support for the intriguing possibility that human micro RNAs translocated into parasite mRNA reduce intra-erythrocytic growth.
Biochemical and mechanical changes in infected HbAS red blood cells have been shown to alter disease progression
Rosette formation was found to be impaired in P. falciparum-infected HbAS red blood cells under deoxygenated conditions]. Impaired rosette formation with HbAS red blood cells may be due to increased sickling of these cells in deoxygenated conditions or to reduced expression of erythrocyte surface adherence proteins . Decreased rosette formation and the resulting decreased circulatory obstruction might contribute to protection against severe malaria in HbAS individuals.
Reduced cytoadherence has also been implicated as a mechanism of protection in HbAS individuals. Infected red blood cells express one of a family of parasite-encoded P. falciparum erythrocyte membrane protein 1 (PfEMP-1) molecules on the erythrocyte surface, and via this protein adhere to endothelial cells in the microvasculature.
Enhanced opsonization and clearance of parasitized HbAS red blood cells by the spleen may lead to increased antigen presentation and earlier development of acquired immunity compared to that in HbAA individuals.
It is likely that both biochemical and immune mechanisms contribute to the protection afforded against falciparum malaria by the HbAS genotype. HbS clearly induces biochemical changes in the red blood cell that may affect parasite metabolism and growth. Plasmodium falciparum also likely inflicts oxidative damage on the HbAS red blood cell. Chronic low levels of oxidized haem in sickled red blood cells may induce HO-1, leading to host tolerance in severe disease.
TREATMENT
Painful vaso-occlusive crisis 1.hydration 2.precepitating factors 3.oxygen therapy 4.analgesic 5.exchange transfusion Antisickling agent (Hydroxyurea). Bone marrow transplantation(BMT).Gene therapy.
maintenance therapy and prevention
1.folic acid 1 mg/d orally.2.pneumococcal vaccine.3.pregnancy (increase crisis, abortion, stillbirth) folate,, exchange transfusion.
4.general anesthesia. Exchange transfusion will increase Hb-A= 60%. Careful hydration and oxygenation.
Angiographic contrast media causing sickling should be avoided
The thalassemia syndromes:
are inherited disorders arising from globin gene mutations that either reduce or totally abolish the synthesis of one or more globin chains. These imbalance in chain synthesis lead to formation of unstable Hb. or decrease Hb. lead to hypochromic microcytic anaemia .The thalassaemia named according to globin chain involved.
Clinical Forms of β-Thalassaemia
Thalassaemia major (TM)
Presents in the first year of life
Subsequently requires regular transfusions and iron chelation to survive
Thalassaemia intermedia (TI)
Presents later in life
May be transfusion independent or require only sporadic transfusions
Pathophysiology of thalassemia :α- thalassemia, gene deletions are responsible for the decrease or absence of α- chains. ß- thalassemia : usually due to an mRNA abnormality. This mutation reduces or eliminates the production of ß-globin chains
Pathogenesis of anaemia of ß-thala.
Decreased B- globin production has two major consequences :total Hb. synthesis is reduced leading to microcytic anaemia, low level of HbA lead to increase Hb-A2,Hb-F. free α-chain accumulates and precipitate in RBC lead to hemolysis, destruction of RBC in the BM.
.Pathophysiologic Sequelae of Untreated Thalassaemia and Corresponding Clinical ManifestationsHaemolysisIneffectiveerythropoiesisMembranebinding ofIgG and C3AnaemiaIncreasederythropoietinsynthesisSkeletaldeformities,osteopaeniaErythroidmarrowexpansionIron overloadSplenomegalyExcess free -globin chainsDenaturationDegradationFormation of haeme and haemichromesIron-mediated toxicityRemoval ofdamaged red cellsIncreased Iron absorptionReduced tissueoxygenation
ThalassaemiaMajor Molecular Basis
Patients with β-thalassaemia major have inherited two β-thalassaemia allelesLocated on each copy of chromosome 11
Hypochromic, abnormally shaped red blood cells
Contain significantly reduced amounts of haemoglobin than normal blood cells because of diminished HbA synthesis
Deposition of precipitated aggregates of free α-globin chains results in accumulation
Damages erythrocytes, precursor cells in bone marrow
Resulting anaemia so severe that patients usually require chronic blood transfusions
Molecular Basis of Thalassaemia Intermedia
3 main reasons
Inheritance of a mild (β+) mutation
Presence of a polymorphism associated with increased HbF
Coinheritance of -thalassaemia
Increase production of alpha-globin chains by
Triplicated alpha genotype associated to beta-heterozygosity
Interaction of beta and delta beta thalassaemia
Epidemiology
Approximately 7% of the world’s population is a carrier of haemoglobin disorders1
Between 300,000 and 500,000 infants are born every year with severe homozygous forms of the disease
An overview of the global distribution of thalassaemias shows that in addition to the Mediterranean countries in which they were first recognized, thalassaemias are frequently found in Asia and the Far East
Population migration has led to spread of this condition with its morbidity and mortality
Clinical Manifestations
Thalassaemia trait has no important clinical effects
Activity of the normal β gene on the allelic chromosome makes enough stable globin
However, inheritance of 2 defective β-globin genes causes a wide spectrum of clinical conditions
Molecular studies reveal a wide array of abnormalities, which underlie above phenotypes and help in their identification
TI has an extraordinarily wide clinical spectrum, unlike TM, which presents with severe anaemia requiring frequent blood transfusionsSevere TIPresentation between 2 and 6 yearsRetarded growth and developmentMild TICompletely asymptomatic until adulthoodThalassaemia IntermediaClinical Features
Clinical manifestations of thalassemia cont>>
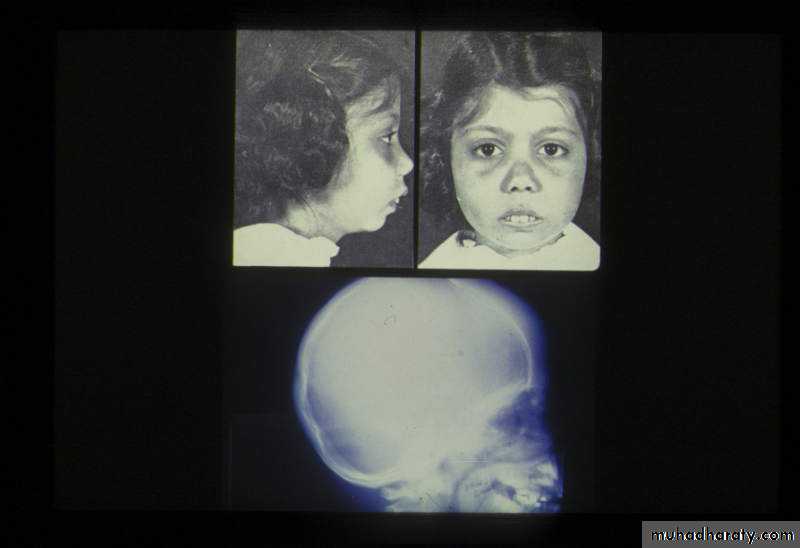
ß- thalassemia major (ßoßo): is the most severe - Becomes apparent 3-6 months after birth when switch from Hb-F to Hb-A takes place :Hepatosplenomegaly(gall stones are also common )Expansion of the bones (hair on end appearance on skull X-ray examination).Severe anaemia with growth retardation and delayed sexual development Damage to heart, pancreas, endocrines and liver due to iron over load

Helpful Clues to Differentiate Major from Intermedia Thalassaemia Major Thalassaemia Intermedia More Likely More LikelyClinical Presentation (years) <2 >2 Hb levels (g/dL) 6–7 8–10 Liver/spleen enlargement Severe Moderate to severeHaematologic HbF (%) >50 10–50 (may be up to 100%) HbA2 (%) <4 >4Genetic Parents Both carriers of high HbA2 1 or both atypical carriers: -thalassaemia - High HbF -thalassaemia - Borderline HbA2Molecular Type of mutation Severe Mild/silent Coinheritance of -thalassaemia No Yes Hereditary persistence of fetal haemoglobin No Yes -thalassaemia No Yes Polymorphism No Yes
DIAGNOSIS OF ß-THALASSAEMIA
Positive family history with:a.non specific findings :Blood smear reveals microcytic RBCs, poikilocytosis, fragmented RBCs, MCV is low (around 65fl).Heinz bodies are evident by supravital stains. b. specific findings :Definitive diagnosis of ß-thala. Is based on the following findings on Hb. Electrophoresis : increased proportion of Hb-A2(> 3.5%). increased proportion of Hb-F.δδαααγγα Hb A2Hb FDiagnosis: β Thalassemia major Genotype αααα - - δδ γγ Haemoglobins ProducedLaboratory diagnosis of β thalassemia major made by CBC, absence of Hb A, with increased Hb F. Some patients have small amounts of Hb A if some β globin chain is produced.
Complications
Thalassaemia major complications mostly due to iron overload and frequent blood transfusions
Heart failure
Infection (blood transfusion, postsplenectomy)
Hypogonadism and infertility
Diabetes mellitus
Hypothyroidism
Thalassaemia intermedia complications include
Thrombosis
Pulmonary hypertension
Leg ulcers
Extramedullary haematopoiesis
Endocrine disorders (osteoporosis, hypogonadism)
Iron Overload
Iron overload occurs when iron intake is increased over a sustained period of time
Transfusion of red blood cells (thalassaemia major)
Increased absorption of iron from the digestive tract (thalassaemia intermedia)
Because there is no mechanism in humans to excrete the excess iron, this has to be removed by chelation therapy
Normal intestinal iron absorption is about 1–1.5 mg/d
In thalassaemic patients who do not receive any transfusion, iron absorption increases
In individuals who are poorly transfused, absorption rises to 3–4 mg/d or more
This represents a supplementary 1–2 g of iron loading per year
1 unit of blood contains approximately 200–250 mg of iron
Chronic transfusion-dependent patients have an iron excess of ~ 0.32–0.64 mg/kg/d
With repeated infusions, iron accumulates
Signs of iron overload can be seen after anywhere from 10 to 20 transfusions, such as in thalassaemia major patients
Iron overload can lead to early mortality
Evaluation of Iron Overload
Serum ferritin concentration
Noninvasive
Accuracy in iron overload questionable
Liver iron concentration (LIC)
Liver biopsy
Reference standard
MRI
Noninvasive, FDA-approved technique
α-THALASSAEMIAS
Decrease synthesis of a-chains, lead to precipitation of Hb-H (4-ß chains) or Hb- Bart’s (4 δ-chains)Classification of α -thalassaemia : Hydrops fetalis : severe, all 4 α -genes are deleted lead to severely anaemic, edematous, stillborn infant. Hb-barts (4 δ -chains had very high oxygen affinity).
Hb-H disease : deletion of 3 α -genes lead to unstable Hb. result in precipitation and extra-vascular hemolysis. α -thalassaemia Trait : deletion of 2 α genes.α -thalassaemia Carrier: deletion of 1 α gene, asymptomatic.DIAGNOSIS OF α-THALA Positive family history with lab finding non specific findings :Blood smear show microcytic, hypochromic red cells, target cells, aniso-poikilocytosis, and decrease MCV.Heinz bodies are evident. Specific findings : definitive diagnosis is finding of Hb-H by Hb elecrophoresis.

NormalControlAbnormalControlAbnormalControlPatient Hb HHbAHbA2
Hb –H Diagnosis
Haemoglobin Electrophoresis:Hb A 91.5 %
Fast moving band 8.5%
Hb A2 and Hb F decreased
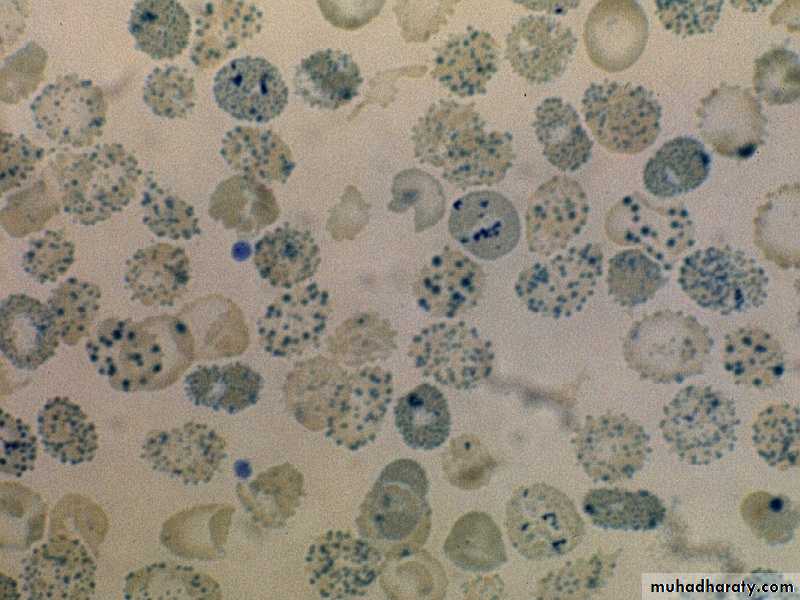
Hb H PreparationHb H inclusions in RBCs
β-Thalassaemia Major Treatment
Conventional treatment/chelation
The gold standard treatment has been the administration of blood transfusions and subsequent iron chelation therapy
Bone marrow transplantation (BMT)
BMT has been attempted from donors with matching alleles
HbF-inducing therapy
Gene therapy—the future
TREATMENT OF α- AND ß- THALASSEMIA 1.Regular red cell transfusions : hyper- transfusion program (keep the level-of-Hb>110g/L)2.Neocytes transfusion (increase RBC survival, decrease requency of transfusion, and decrease iron over load). 2-3 units every 4-6 weeks .3.Leukocyte filter will lowers rate of transfusion reaction.4.Folic acid supplementation (5 mg) to prevent aplastic crisis.5.Iron chelation:Desferioxamine (Desferal) either with each unit of transfused blood (2 g) or by slow subcutaneous daily infusions by pump (1-4g.over(8-12) (Exjade)Deferasirox.Deferiprone 6.Splenectomy : mechanical difficulty, hypersplenism.7.BMT : prior to development of hepatomegaly, portal fibrosis & iron over load.
PRENATAL DIAGNOSIS OF THALASSAEMIA
Guide parents and physicians in deciding whether to complete pregnancy.Both parent carriers. Fetal diagnosis:fetoscope to sample fetal venous blood show α/ß chain synthesis ratio.Amniocentesis or trophoblast (chorionic villus) biopsy for DAN analysis using DNA probes.
By : younis alomary