INTERSEX
INTERSEXDisorders of sexual development (DSD), formerly termed intersex conditions, are among the most fascinating conditions encountered by the clinician. The ability to diagnose these conditions has advanced rapidly in recent years. In most cases today, clinicians can promptly make an accurate diagnosis and counsel parents on therapeutic options.
“Intersex” is a general term used for a variety of conditions in which a person is born with a reproductive or sexual anatomy that doesn’t seem to fit the typical definitions of female or male.
INTERSEX
Intersex covers a diverse range of conditions encompassing:individuals with standard male or female genitalia, who may have a variety of internal genital organs and karyotypes, and
also those with ambiguous external genitalia.
For example, a person might be born appearing to be female on the outside, but having mostly male-typical anatomy on the inside.
Or a person may be born with genitals that seem to be in-between the usual male and female types—for example, a girl may be born with a noticeably large clitoris, or lacking a vaginal opening, or a boy may be born with a notably small penis, or with a scrotum that is divided so that it has formed more like labia.
Or a person may be born with mosaic genetics, so that some of her cells have XX chromosomes and some of them have XY.
#CLASSIFICATION OF INTERSEXUALITY
Virilization of genetically female foetusFemale pseudohemaphroditism
Incomplete musculinization of genetically male foetus
Male pseudohermaphroditism (XY-FEMALE)
The presence of both ovarian and testicular tissue in the same individual True hermaphroditism
Chromosomal abnormality
Mixed gonadal dysgenesis ( 45,X0 / 46,XY)
This terminology mainly reflects the chromosomal sex or the gonadal tissue associated with the disorder.
How many children are born with intersex conditions?
A conservative estimate is that 1 in 2000 children born will be affected by an intersex condition.
#Aetiology
Most intersex conditions occur due to a genetic or environmental disruption to the pathway of fetal sexual development. This disruption can be to:Gonadal differentiation or development,
Sex steroid production,
Sex steroid conversion,
Tissue utilization of sex steroid.
Male pseudohermaphroditism (XY- FEMALE)
Failure to produce testosteroneAnatomical testicular failure (Pure XY gonadal dysgenesis (swyer’s syndrome), testicular regression syndrome)
Leydig-cell agenesis
Enzymatic testicular failure
Failure to utilize testosterone
5-alpha-reductase deficiency
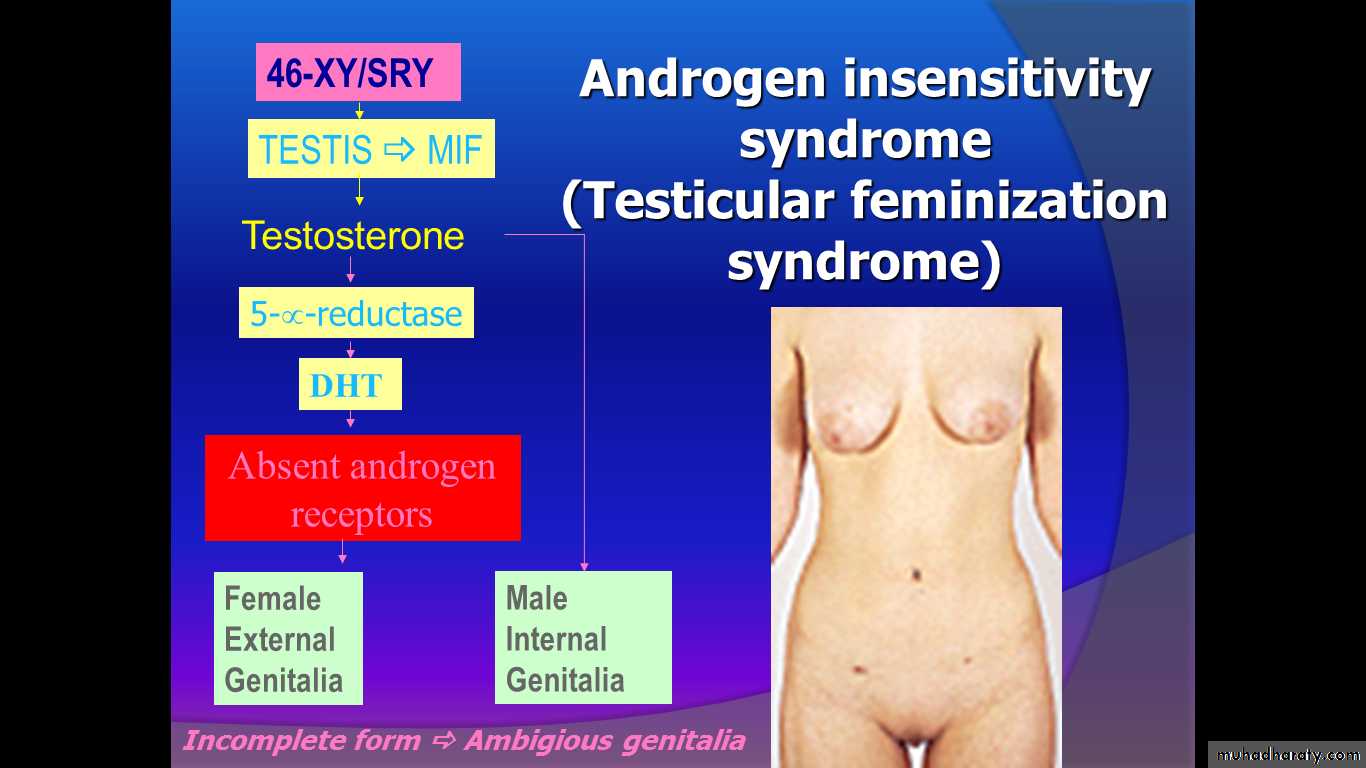
Androgen receptor deficiency
* Complete androgen insensitivity (TFS)
* Incomplete androgen insensitivity
Anatomical testicular failure:
Failure of testicular differentiation and development result from sex chromosomes mosaicism or may be associated with normal chromosomes in pure gonadal dysgenesis.
These patient have poor musculinization or none,
uterus, tubes and vagina are present (in contrast to other XY females).
#Swyer’s syndrome
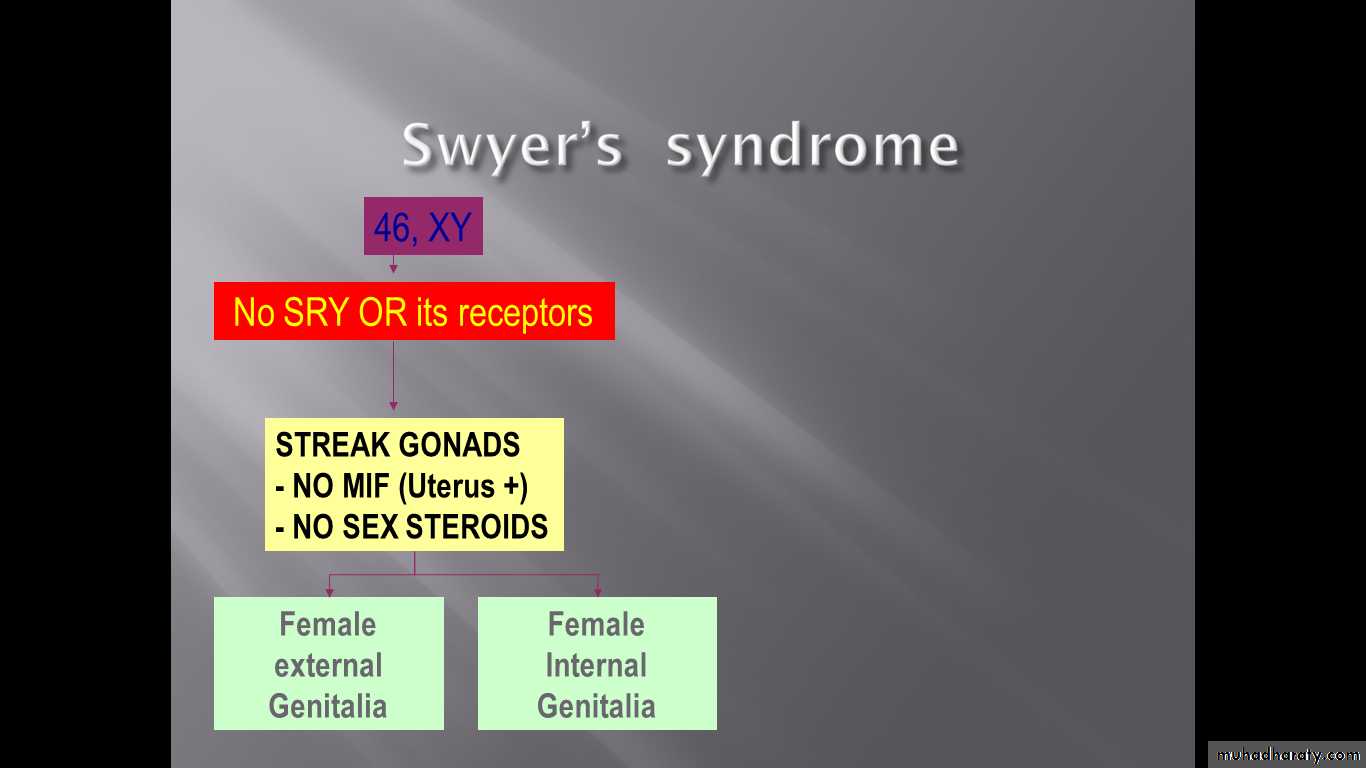
#Treatment :-removal of streak gonads during childhood (risk of malignancy in 5%)-puberty replacement therapy (oestrogen and progesterone) to produce secondary sexual development and menstruation

Enzymatic testicular failure:
Many biosynthetic defect in the formation oftestosterone from cholesterol, usually the defect
is incomplete so there is external genital ambiguity of various degree ,the uterus ,upper vagina are absent because of production of the MIF by the testes is normal .
The chosen sex is often female depend on external organ suitability .surgery may be used as previously described.

5 alpha reductase deficiency
This enzyme deficiency is autosomal recessive so family history of such cases may be present. It affect the conversion of testosterone to DHT required for masculinization of the ext. genetalia in the male (poor musculinization so placed in female role) ,there is normal production of MIF so always there is absence of the uterus and vagina.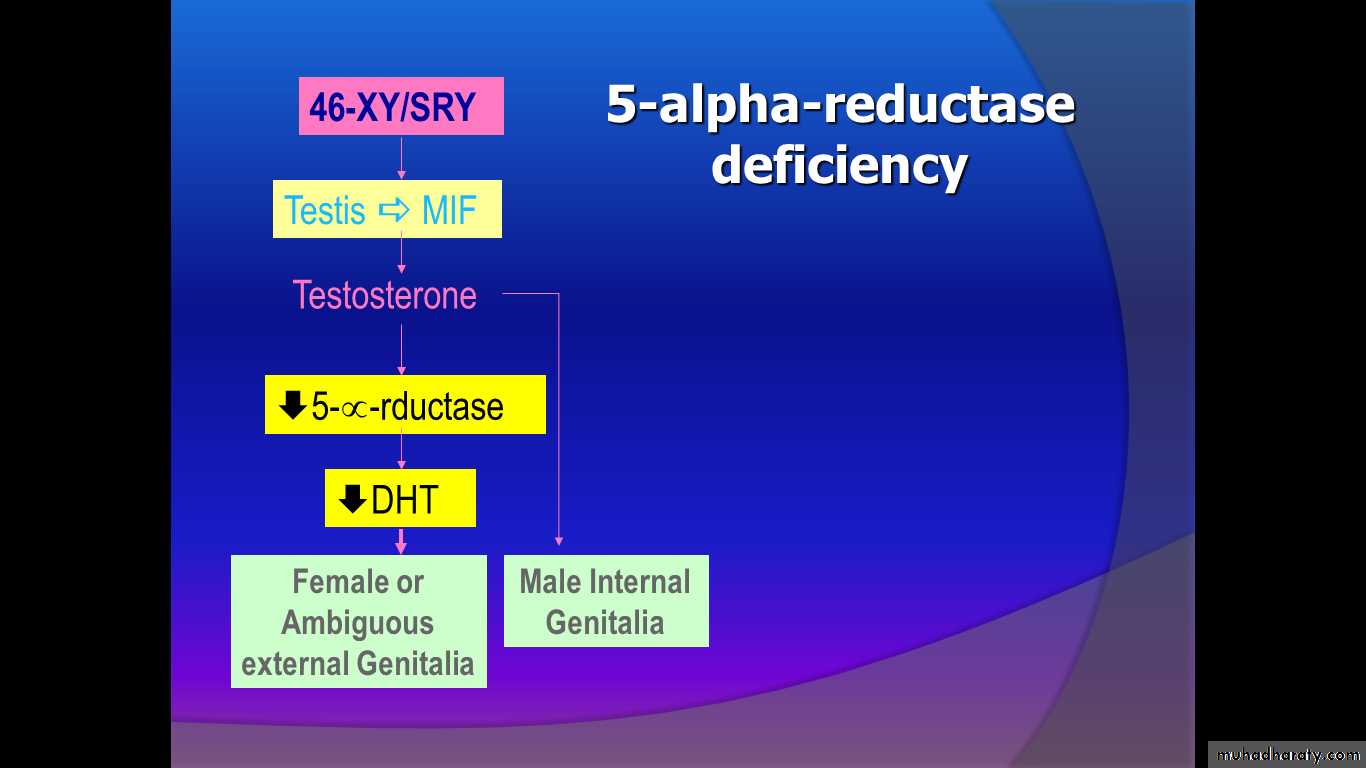
#Clinical features:
Female phenotype.primary amenorrhoea.
Normal breast development .
Scanty or absent pubic and axillary hair.
Absent uterus and tubes with short vagina.
Undescended or maldescended testes (found in the abdomen ,groin or labia majora).
Testosterone and oestrogen level are in normal male range.
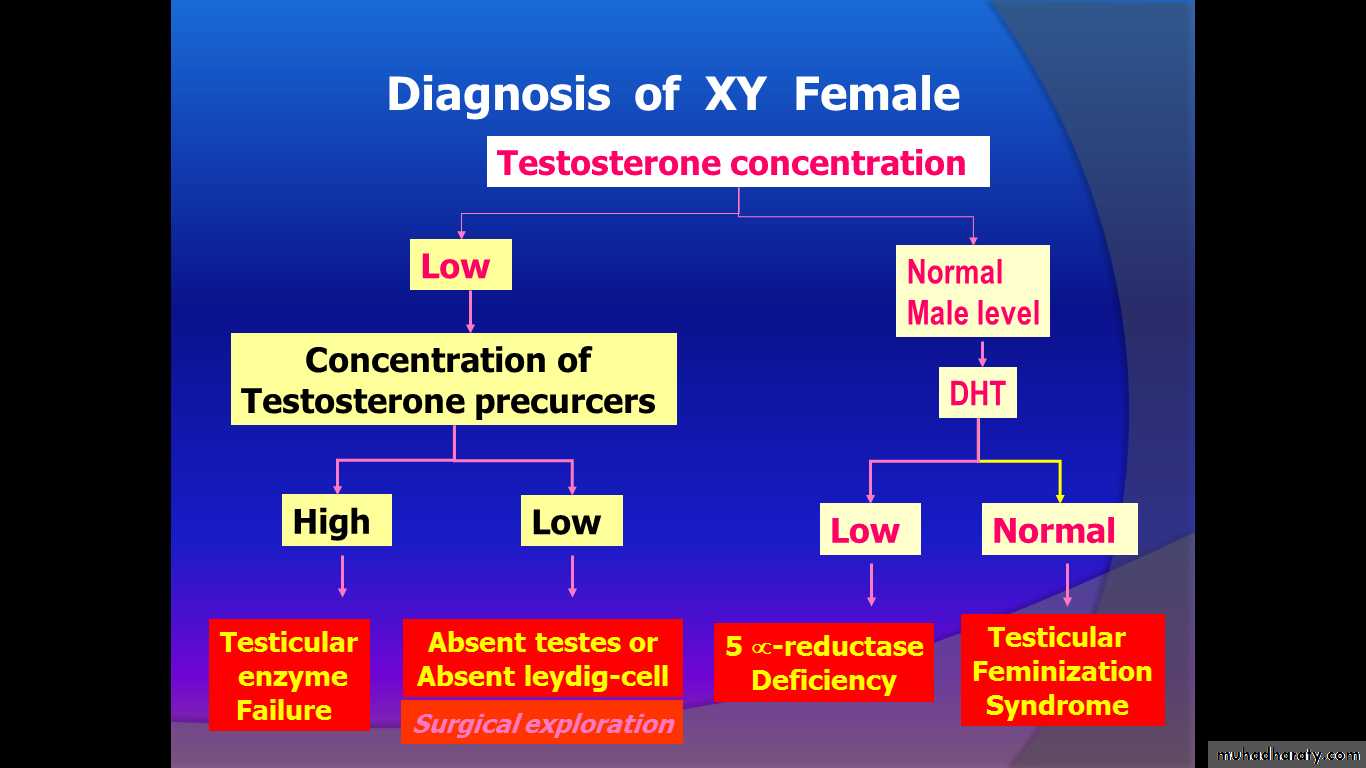
Diagnosis: female appearance +46XY.
Emphasize that these are entirely female despite their 46XY karyotype.
5% risk of malignancy require gonadectomy.
Oestrogen (HRT) treatment may be required.
Surgery to elongate the vagina is seldom required.
True hermaphrodite:
The presence of both ovarian tissue containing grafian follicle and testicular tissue containing distinct tubules in one person.These patients presents with various degree of
sexual ambiguity (from maleness to femaleness)
In the majority karyotype of 46XX(58%), other
less common as 46XX/XY, 46XY and 46XY/XXY.
Regarding gonadal distribution ovotestis present
on one side and ovary on the other or ovary in
one side and testis on the other (two most frequent).
In the majority uterus and vagina are present.
Diagnosis by biopsy. (Gonadal biopsy is not under taken to determine the sex of rearing which should be undertaken depending on the suitability of the external genitalia for sexual life )
The sex chosen depend on functional capability of external genitalia .
inappropriate organ should be removed.
#FEMALE PSEUDOHERMAPHRODITISM
EXCESS FETAL ANDROGENS
Congenital adrenal hyperplasia21 -hydrxylase deficiency
11-hydroxylase deficiency
3ß-hydroxysteroid
dehydrogenase deficiency
EXCESS MATERNAL ANDROGENS
Maternal androgen secreting tumours (ovary, adrenal)Maternal ingestion of androgenic drugs
#Congenital Adrenal Hyperplasia(CAH)
Is the most common cause of female intersex (constituting approximately 60% of all intersex cases) & is the most frequent cause of ambiguous genitalia in the newborn.
Autosomal recessive disorder of enzyme deficiency in the cortisol biosynthesis .
Commonest (90%): 21-hydroxylase deficiency
Less common : 3β-hydroxy steroid dehydrogenase deficiency
11β-hydroxylase deficiency
Pathophysiology
The term congenital adrenal hyperplasia (CAH) describes several autosomal recessive disorders that result from complete or partial deficiency of an enzyme involved in cortisol and aldosterone synthesis, usually 21-hydroxylase or less frequently 11-hydroxylase.
Symptoms of CAH and their severity are varied. It may present in the neonate with ambiguous genitalia and life threatening hypotension.
Alternatively, symptoms may be milder and delayed until adolescence or adulthood. In this late-onset form of CAH, symptoms reflect accumulation of precursor C19 steroid hormones. These precursors are converted to dehydroepiandrosterone, androstenedione, and testosterone, and signs of virilization predominate.
The basic biochemical defect is an enzymatic block that prevents sufficient cortisol production. Biofeedback via the pituitary gland causes the precursor to accumulate above the block. Clinical manifestation of CAH depends on which enzymatic defect is present.
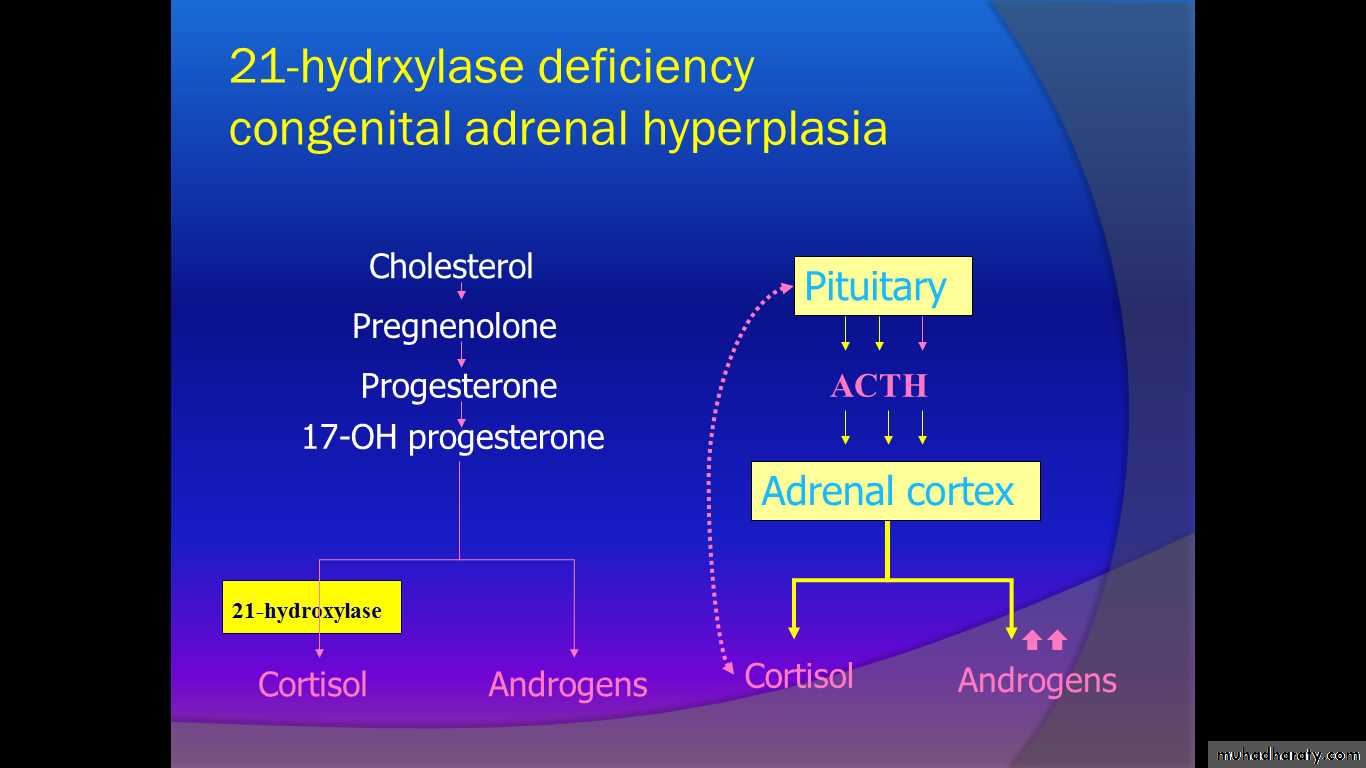
Severe forms of congenital adrenal hyperplasia are potentially fatal if unrecognized and untreated because of the severe cortisol and aldosterone deficiencies that result in salt wasting, hyponatremia, hyperkalemia, dehydration, and hypotension.
Epidemiology
Typically due to a 21-hydroxylase deficiency, classic congenital adrenal hyperplasia (CAH) is one of the most common autosomal recessive metabolic diseases, estimated to occur in 1:10,000 to 1:15,000 births.Congenital adrenal hyperplasia caused by 11-beta-hydroxylase deficiency accounts for 5-8% of all congenital adrenal hyperplasia cases.
Although CAH has been reported in a wide range of ethnic groups, it is most common in the Ashkenazi Jewish population.
Clinical Presentation
Sex:
Because all forms of congenital adrenal hyperplasia are autosomal recessive disorders, both sexes are affected with equal frequency. However, because accumulated precursor hormones or associated impaired testosterone synthesis impacts sexual differentiation, the phenotypic consequences of mutations or deletions of a particular gene differ between the sexes.
Age
Classic congenital adrenal hyperplasia is generally recognized at birth or in early childhood because of ambiguous genitalia, salt wasting, or early virilization.
Nonclassic adrenal hyperplasia is generally recognized at or after puberty because of oligomenorrhea or virilizing signs in females.
History:
The clinical phenotype of congenital adrenal hyperplasia depends on the nature and severity of the enzyme deficiency.Although the information below is presented according to chromosomal sex, the sex of a neonate with congenital adrenal hyperplasia is often initially unclear because of genital ambiguity.
Clinical presentation in females
Females with severe forms of adrenal hyperplasia due to deficiencies of 21-hydroxylase, 11-beta-hydroxylase or 3-beta-hydroxysteroid dehydrogenase have ambiguous genitalia at birth due to excess adrenal androgen production in utero. This is often called classic virilizing adrenal hyperplasia.
affected females born with:
-Enlargement of the clitoris-Excessive fusion of the genital fold obscure the vagina and urethra
-Thickening , rugosity of the labia majora
-Internal genital organ are present.
-dangerous salt losing syndrome may cause death if not treated
In a nonclassic form of CAH, also known as late-onset or adult-onset CAH, hyperandrogenemia does not present until puberty.
At puberty, activation of the adrenal axis increases steroidogenesis, unmasking a mild 21-hydroxylase activity deficiency. Levels of ACTH may increase due to the lack of negative feedback by cortisol, further exacerbating androgen production.
These patients often present with hirsutism, acne, and anovulation. Thus, late-onset CAH may mimic polycystic ovarian syndrome (PCOS).
Females with 17-hydroxylase deficiency appear phenotypically female at birth but do not develop breasts or menstruate in adolescence because of inadequate estradiol production. They may present with hypertension.
Clinical presentation in males
21-hydroxylase deficiency in males is generally not identified in the neonatal period because the genitalia are normal. If the defect is severe and results in salt wasting, these male neonates present at age 1-4 weeks with failure to thrive, recurrent vomiting, dehydration, hypotension, hyponatremia, hyperkalemia, and shock (classic salt-wasting adrenal hyperplasia).Patients with less severe deficiencies of 21-hydroxylase present later in childhood because of the early development of pubic hair, phallic enlargement, or both, accompanied by accelerated linear growth and advancement of skeletal maturation (simple virilizing adrenal hyperplasia).
In male infants, the disease may be misdiagnosed as gastroenteritis or pyloric stenosis, with potentially disastrous consequences due to delayed treatment with glucocorticoids.
Investigation for suspected CAH:
-Karyotype-17 alpha hydroxyprogesterone measurement
-Electrolyte abnormality : Na, Cl , K.
-Pelvic ultrasound (internal genital organ)
Treatment (CAH):
Medical control of the underlying disorder:
Cortisol administration
Correction of electrolyte disorder
Surgical correction of the underlying anatomical abnormality
with two type of surgery :
These patients are genetic females, potentially fertile and must brought up in the female role regardless of the degree of musculinization.
-reduction of the clitoris :amputation or reduction
clitoroplasty (best in the neonatal period)
-division of the fused labial fold (if thick better done well
after puberty)
Prognosis for CAH:
Delayed puberty up to 2 yearsmenstrual abnormality .. oligomenorrhoea to amenorrhoea.
Reduced fertility.
#Congenital adrenal hyperplasia
The commonest cause of genital ambiguity at birth21-Ohase deficiency is most common form
Autosomal reccessive
Salt wasting form may be lethal in neonates
SERUM 17OH-progesterone (21OHase)
SERUM deoxycorticosterone, 11-deoxycotisol (11- OHase)
Treatment : cortisol replacement and ? Surgery
#AMBIGUOUS GENITALIA AT BIRTH
The external genital organs look unusual, making it impossible to identify the sex of the newborn from its outward appearance.
Ambiguous genitalia represent a social and potential medical urgency that is best handled by a team of specialists, which may include urologists, neonatologists, endocrinologists, and pediatric gynecologists.
Child with ambiguous genitalia may be any of the following:
- Musculinized female due to congenital adrenal hyperplasia (CAH), or androgen stimulation from other source.- undermusculinized male.
- True hermaphrodite.
#MANAGEMENT OF NEWBORN WITH AMBIGUOUS GENITALIA
GENERAL GIUDELINES
Medical and social emergency
Avoid immediate declaration of sex
Proper counselling of the parents
Team management; obstetrician, neonatologist, pediatric endocrinolgist, genetist and paediatric surgeon.
The first questions parents ask after a baby is born include, “Is it a boy or a girl?” In the case of ambiguous genitalia, the parents should be informed that the baby's genitals are not fully developed and, therefore, a simple examination of the external genitalia cannot determine the actual sex. The parents should be told that data will be collected but that it may take several days or longer to determine the baby's intended sex. In some situations, it may be best to state simply that the baby has some serious medical complications. The issues of sex assignment and timing of surgical therapy are controversial and should be managed by clinicians with extensive experience in this area.
#MANAGEMENT OF NEWBORN WITH AMBIGUOUS GENITALIA
History : pregnancy; familyDetailed examination : abdomen; pelvis; external genitalia; urethral and anal openings.
Federman’s rule: a palpable gonad below the inguinal ligament is testes until proven otherwise
Investigations :
• Rule out cong. Adrenal hyperplasia: Serum electrolytes; 17-OHP level and urinary levels of 17-ketosteroids
• Karyotype ( buccal smear; blood)
• Pelvic US and sometimes MRI
• Skin biopsy; fibroblast culture to measure 5alpha-reductase activity or dihydrotestosterone binding
• Laparoscopy
• Gonadal biopsy (laparotomy)

SURGICAL CONSIDERATIONS
Phallic / clitoral reduction if the assigned sex is female, before 3 years of ageRemoval of intra-abdominal gonads / streaks in newborns carrying Y chromosome
Vaginal construction / repair is better performed around puberty
INTERSEXUALITY PRESENTING AT ADOLESCENCE
Primary amenorrhea- Complete androgen insesitivity (TFS)
- Congenital anorchia
( early testicular regression syndrome)
- Complete leydig-cell agenesis
- Some forms of enzymatic testicular failure
Ambiguous genitalia
- Neglected congenital adrenal hyperplasia
- Mixed gonadal dysgenesis
- Partial androgen resistance
- Congenital anorchia ( Late )
- Testicular enzymatic failure
- Leydig cell agenesis
( incomplete)
- True hermaphrotidism
Transsexualism
Brain sex: cerebral differentiation to a male or female orientation.Transsexualism occurs when a person strongly believes that he or she belong to the opposite sex.
This is typically a lifelong feeling and results in varied degrees of physical/external changes
These patients should be referred to the psychiatrist
Other chromosomal abnormalities of sexual developement
Klinefelter syndrome XXY +ve Bar body male+medical problemTurner syndrome Xo -ve Bar body
female + medical problem
super female XXX two+ve Bar body
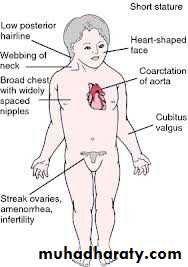
Turner syndrome:
45XO karyotype.
Present as delayed puberty with short stature.
Absent secondary sexual characteristics.
Wide carrying angle of the arm.
Webbed neck.
broad chest with widely spaced nipples.
Associated medical problems as colour blindness, coarctation of the aorta.
Streak ovaries (non functioning).
Normal internal genital organs.
Markedly elevated gonadotrophin (LH,FSH)
#Treatment :
Induction of puberty by long treatment of oestrogen.Induction of menstruation by adding progesterone to oestrogen after 2 years of oestrogen treatment.
If karyotype reveal 46XX(gonadal dysgenesis)
the gonads have 30% risk of malignant change
KLINEFILTER SYNDROME:
Appearance essentially is male.
Scrotal testis ,small azoospermic (infertile).
Small penis with normal potency.
Karyotype of 47XXY(chromatin positive) rare as 48 XXXY ,mosaics of XX and XY.
Super female:
Karyotype is XXXmental subnormality
hypoplastic genitalia
Scanty menstruation or amenorrhoea