Fifth stage
PEDIATRIC SURGERYLec-
د.عبدالرحمن
9/11/2015
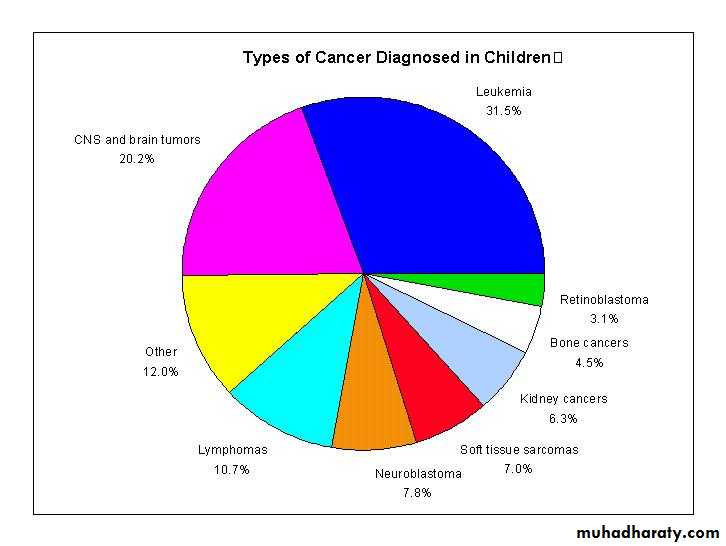
PEDIATRIC ONCOLOGYLeukemia is the most common childhood cancer
Brain tumors are second most common
Lymphomas are the third most common
Then solid tumors outside the CNS
Neuroblastoma - neural crest derived
Wilms - renal tumors and syndromes
Bone tumors
Rhabdomyosarcoma - soft tissue sarcomas
Sacrococcygeal teratoma
Lymphoma
LymphomaLeukemia
Leukemia
Brain Tumors
Brain Tumors
Leukemias
Definition and General Characteristics Uncontrolled proliferation of immature white blood cells with a different immunological
subtype which is lethal within 1–6 months without treatment
The disorder starts in the bone marrow, where normal blood cells are replaced by
leukemic cells
Morphological, immunological, cytogenetic, biochemical, and molecular genetic
factors characterize the subtypes with various responses to treatment
Leukemia: Signs and Symptoms
Bone marrow infiltrationAnemia
Pallor, lethargy
Dyspnea, murmur
Platelets
Bleeding, petechiae, purpura
Neutropenia
Fevers and infections
Bone pain
Limp, walking, irritability
Extramedullary spread
LymphadenopathyHepatosplenomegaly
Orthopnea, cough
mediastinal mass
tracheal compression
Facial nerve palsy
Testicular enlargement
Skin lesions
Gingival hypertrophy
Fever of malignancy
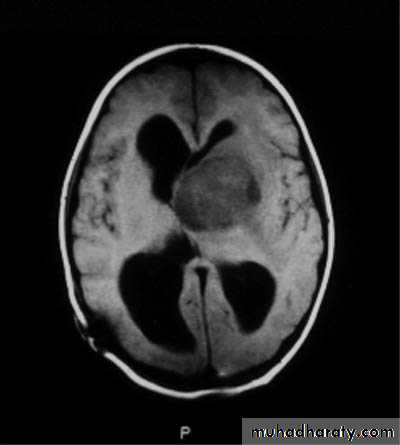
CNS Tumors
MRI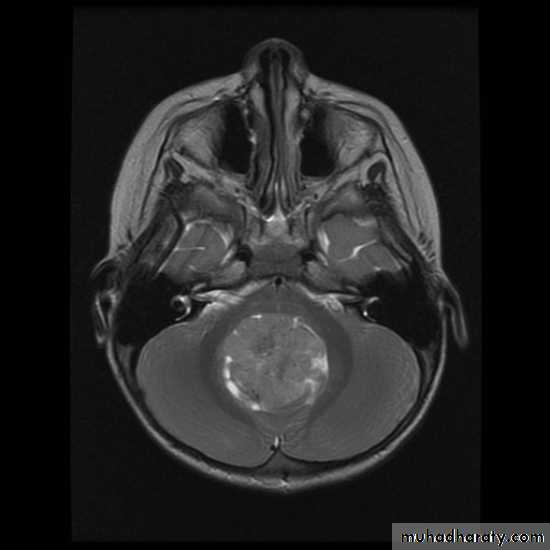
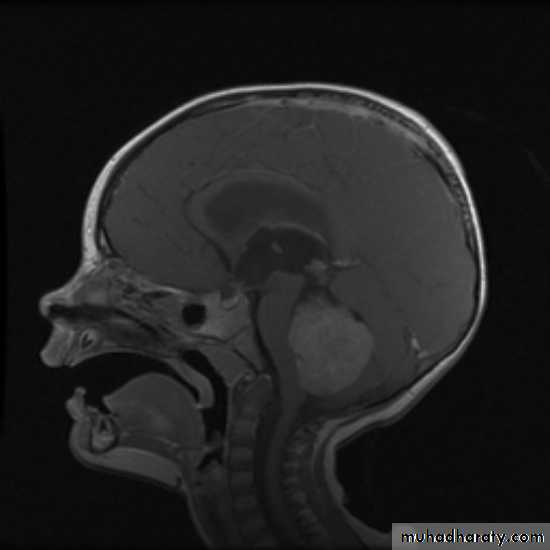
Brain Tumors of Childhood
Histogenesis:* Cell of origin:
glial, neural, primitive, choroid, mixed
* Location:
posterior fossa: 70%
supratentorial: 30%
* Clinical presentation:
location
age
type and grade of the tumor
Brain Tumors of Childhood
Infratentorial70%
esp. < 6 y/o
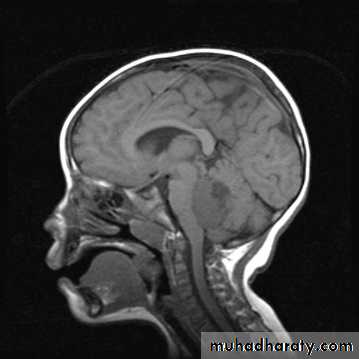
Supratentorial
30%esp. > 8 y/o
Symptoms may include:
Increased intracranial pressuresecondary to obstruction of CSF at aqueduct
hydrocephalus (infants), headache, papilledema, vomiting
seizures
focal neurological deficits
hormonal changes (pituitary adenoma)
visual changes (diplopia, field defects)
pressure on optic chiasm
Lymphomas
Childhood LymphomasSigns and Symptoms depend on:
Lymphoma subtype
Hodgkin’s Disease (HD)
Non Hodgkin’s Lymphoma (NHL)
* Lymphoblastic
* Burkitt’s
* Large Cell lymphoma
Location
Presentation of Hodgkin’s Disease
Age: adolescents >> young child
Painless lymphadenopathy
Progresses over weeks months
Location
95%
95%
Cervical/supraclavicular LNS
unilateral or bilateral
Mediastinal ± hilum
LNs below diaphragm and spleen
Liver, lung, bone marrow

Systemic symptoms
“B” symptoms25%
“B” symptoms
25%
Fevers
Night sweats
Weight loss
Pruritus
Superior Mediastinal Syndrome (SMS)
Orthopnea, SOB, strider, hypoxia
compression
compression
Tracheal
= Oncologic Emergency
= Oncologic Emergency
Bronchial
Cardiac
Presentation of Non Hodgkin's lymphoma
Lymphoblastic lymphoma
Burkitt’s Lymphoma
B-cell origin> 5 y/o
Abdominal mass
Large mass + LNs
Terminal ileum, Cecum or appendix
Jaw
Tumor lysis syndrome
Uric acid, phosphorus, creatinine
Treatment can precipitate renal failure
= Oncologic Emergency
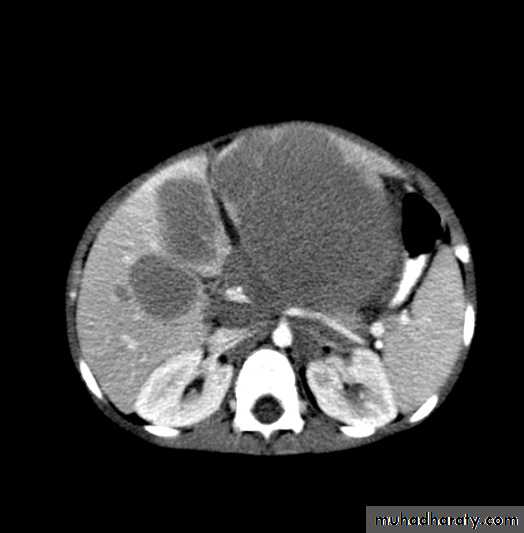
Other Abdominal Tumors
Malignant Abdominal MassesMost common:
Burkitt’s lymphoma
Neuroblastoma
Wilms Tumor
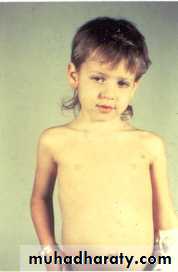
Other:
HepatoblastomaRhabdomyosarcoma
pelvic
Ovarian germ cell tumors
pelvic
Neuroblastoma
Age90% < 5 y/o; 50% < 2 y/o
Occasional ultrasonography detection in utero
Location: any neural crest tissue
Adrenal
Paraspinal sympathetic tissue
Cervical, Thoracic, Pelvic
Often metastatic at diagnosis
Bone and/or bone marrow
Neuroblastoma: Signs and Symptoms
Abdominal mass
Often crosses midline
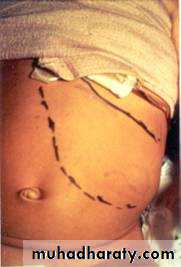
Lower extremity weakness
Spinal cord compressionThoracic
abdominal
Cervical, high thoracic mass
Horner’s syndrome
Miosis, ptosis, anhydrosis
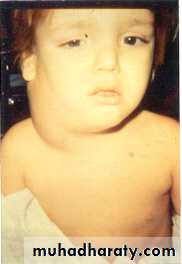
Signs of metastatic disease
IrritabilityBone pain

Fever
Proptosis
Bone lesions
Periorbital
ecchymoses
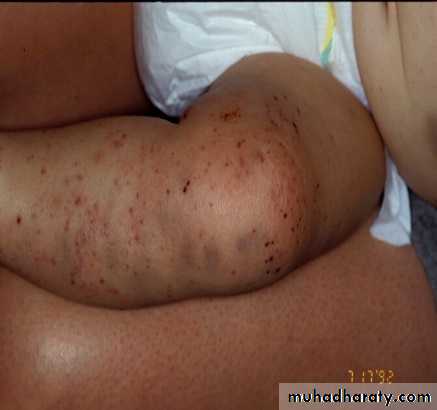
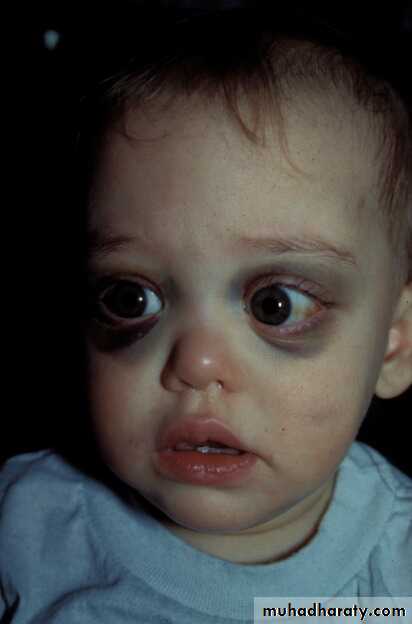
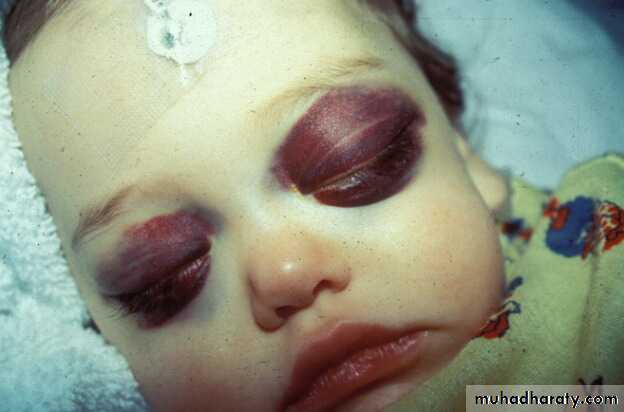
Periorbital Ecchymoses of Neuroblastoma
1 month into
therapy
1 month into
therapy
13 months old
at diagnosis
13 months old
at diagnosis
Paraneoplastic syndromes
Watery diarrhea – Vasoactive Intestinal PeptideUrinary catecholamines

VMA – 85%
BP – 25%
Renal compression
Catecholamine secretion
Wilms tumor: Signs and Symptoms
Abdominal mass
Often asymptomatic
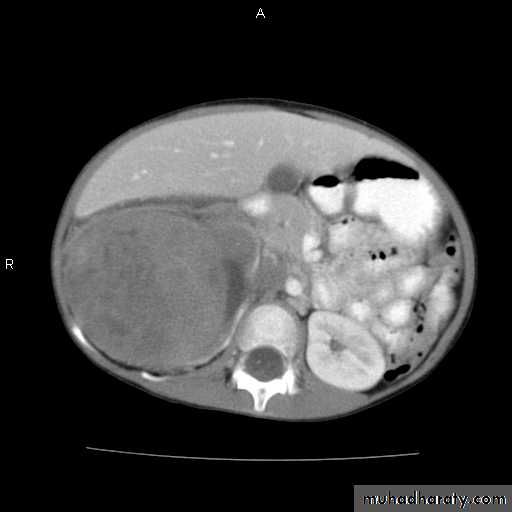
Encapsulated
mass
Encapsulated
mass
Healthy appearing

2 days
before
dx
2 days
before
dx
BP – 25%
Mass enlarges toward pelvis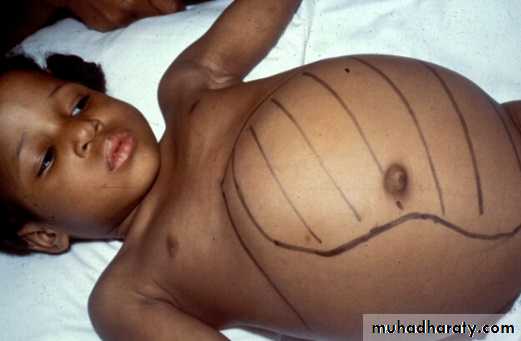
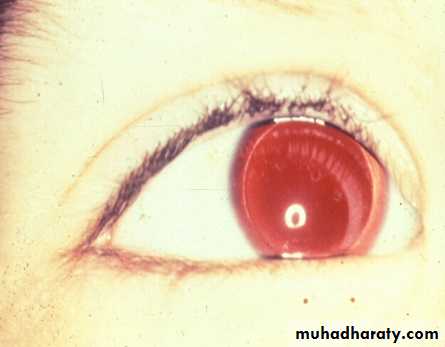
Associated anomalies, syndromes – 15%
WAGR syndromeWilms, aniridia, ambiguous genitalia, mental retardation
due to 11p13 deletion (WT-1)
Aniridia
Bone tumors
Bone Tumors in ChildhoodAge – Adolescents > younger children
Signs and symptoms
Bone pain, ± palpable mass, ± ¯ motion
Often hx of sports injury (coincidental)
Ewing Sarcoma
All bones:
Long: diaphyses
Flat
Pelvis
Skull
Ribs
Ewing Sarcoma
All bones:
Long: diaphyses
Flat
Pelvis
Skull
Ribs
Osteogenic Sarcoma
Metaphyses of long bones:
Distal femur
Proximal tibia
Proximal humerus
Pelvis
Osteogenic Sarcoma
Metaphyses of long bones:
Distal femur
Proximal tibia
Proximal humerus
Pelvis
Presentation of Bone Tumors
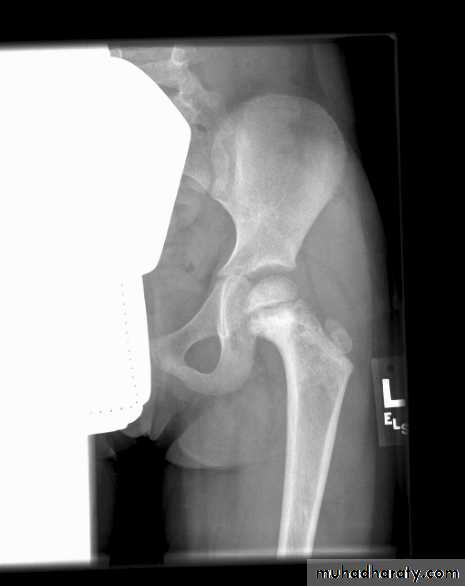
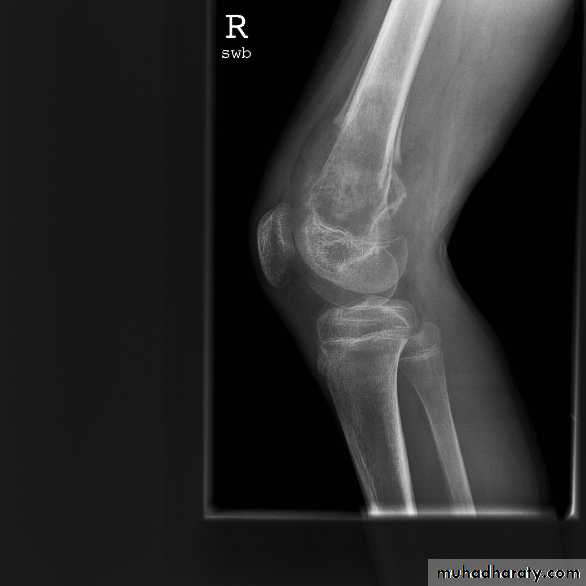
Plain X-Rays are usually abnormalClassic X-ray of Ewing:
Moth-eaten
lytic lesion
Classic X-ray of Ewing:
Moth-eaten
lytic lesion
Classic X-ray of O.S.:
“Sunburst pattern”Periosteal reaction
Soft tissue mass + calcium
Classic X-ray of O.S.:
“Sunburst pattern”
Periosteal reaction
Soft tissue mass + calcium
Presentation of Bone Tumors
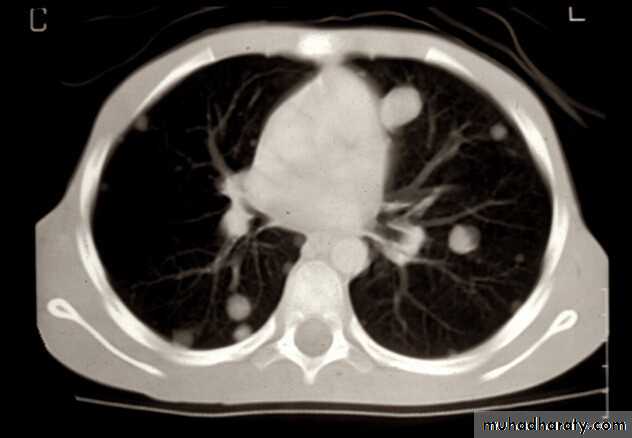
Further radiographic evaluation may help with differential diagnosis of bone painBone scan
MRI
Chest CT scan
Metastases 20%
Soft tissue sarcomas
Presentation of Soft Tissue SarcomasRhabdomyosarcoma – most common
Age
Birth to > 20 y/o
70% < 10 y/o
Sites
Head and neck – 40%
Signs and symptoms
depend on
age and site
Signs and symptoms
depend on
age and site
Genitourinary – 20%
Extremities – 20%
Trunk – 10%
Retroperitoneal – 10%
Rhabdomyosarcomas: Signs and Symptoms
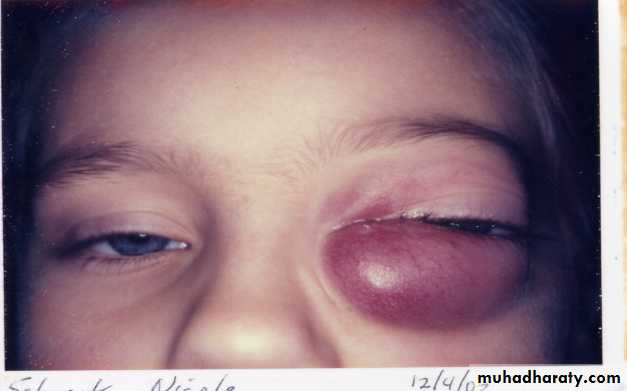
Head and neck
Orbit
Proptosis
Periorbital swelling
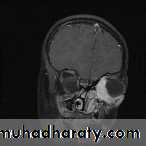
Parameningeal
Cranial nerve palsies
Hearing loss
Chronic aural or sinus drainage
Rhabdomyosarcomas: Signs and Symptoms
Genitourinary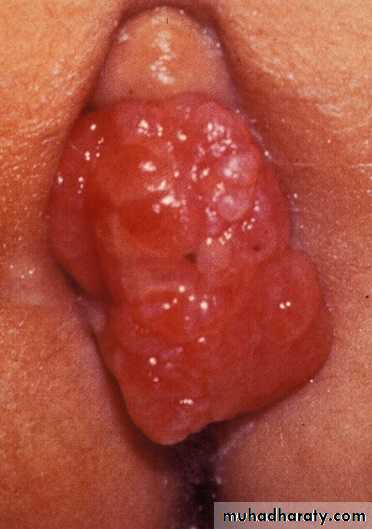
Botryoid:
grape-like
Botryoid:
grape-like
Bladder and prostate
Hematuria
Urinary obstruction
Paratesticular
Painless mass - testicle
Vagina and uterus
Abdominal mass
Vaginal mass
Vaginal bleeding or discharge
Rhabdomyosarcoma – other sites
Can grow up at any site and any age
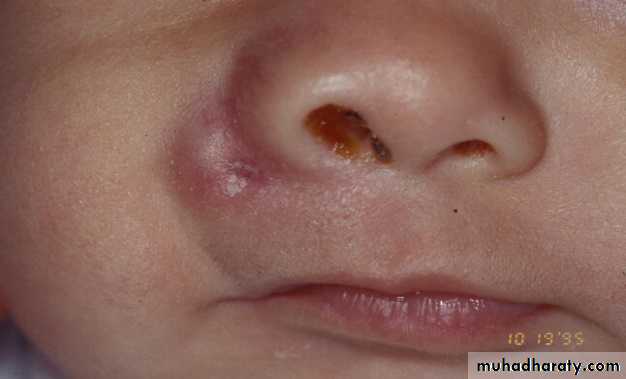
6 week old
6 week old
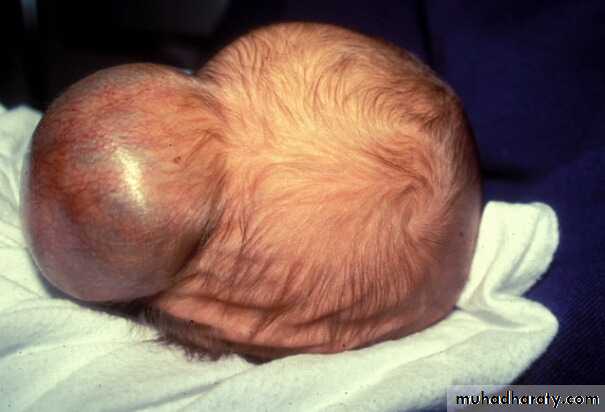
Newborn
Newborn
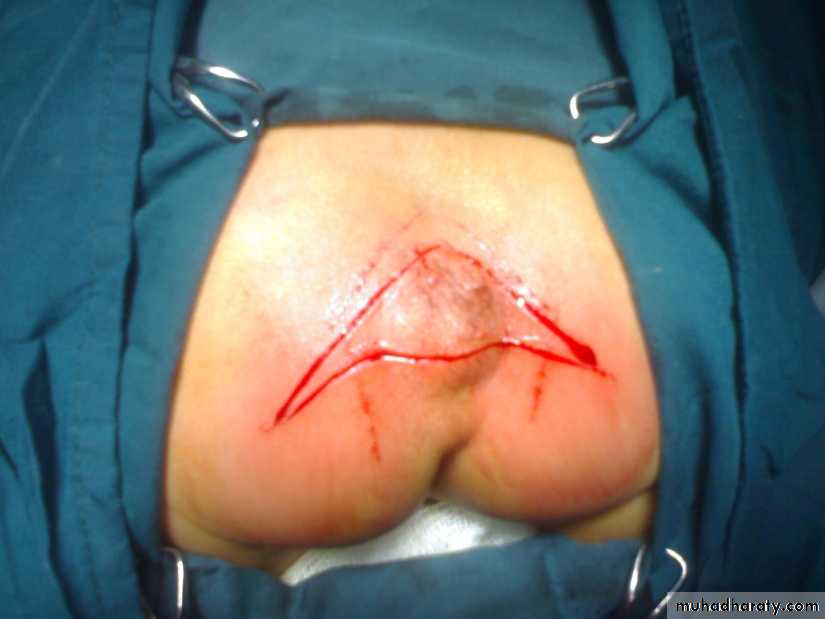
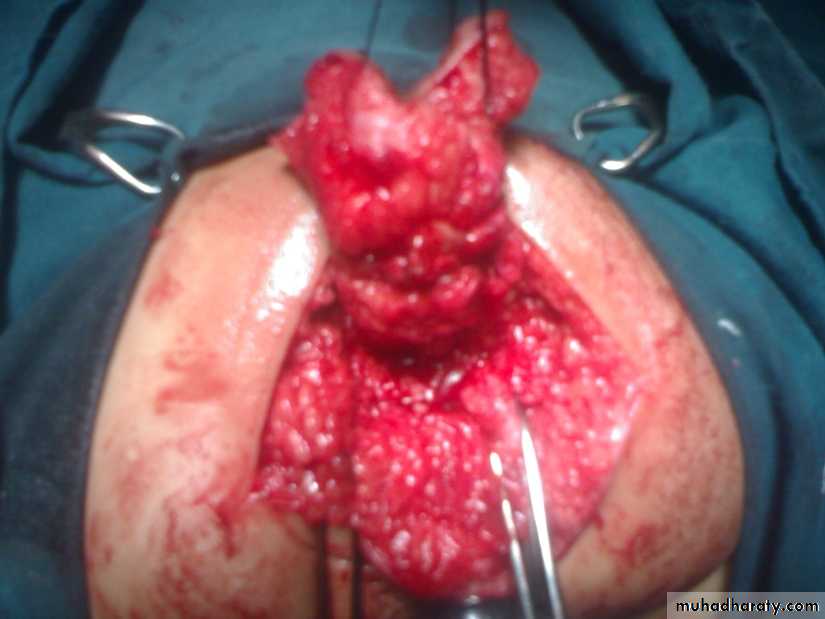
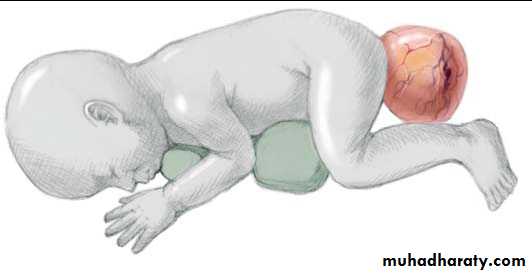
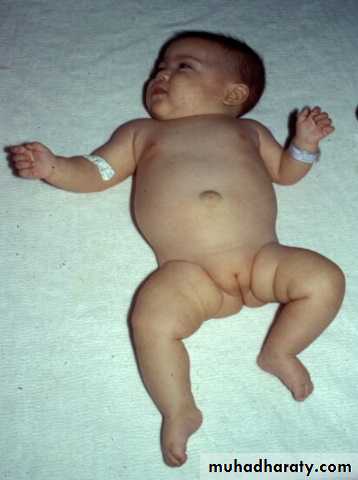
Sacrococcygeal teratoma
Its a rare tumor, occurring in approximately 1 in 40,000 live births. They arise from the caudal end of the spine, usually protrudingfrom the inferior end of the infant’s spinal column and displacing the anus forwards. They are much more common in girls, with the female to male ratio being at least 3:1. It is generally agreed that sacrococcygeal teratoma is the result of continued multiplication of totipotent cells from Hensen’s node, which fail to involute at the end of embryonic life.