
1
Fifth stage
Medicine
Lec-15
د.خالد نافع
25/4/2016
HEMOGLOBINOPATHIES
Definition:
Hemoglobinopathies are inherited disorders in which Mutation in or near the globin genes
alter the structure of amino acid sequences or the rate of synthesis of a particular globin
chain.
Hb-S B6 glu val
Hb-C B6 glu lys
Defective hemoglobin
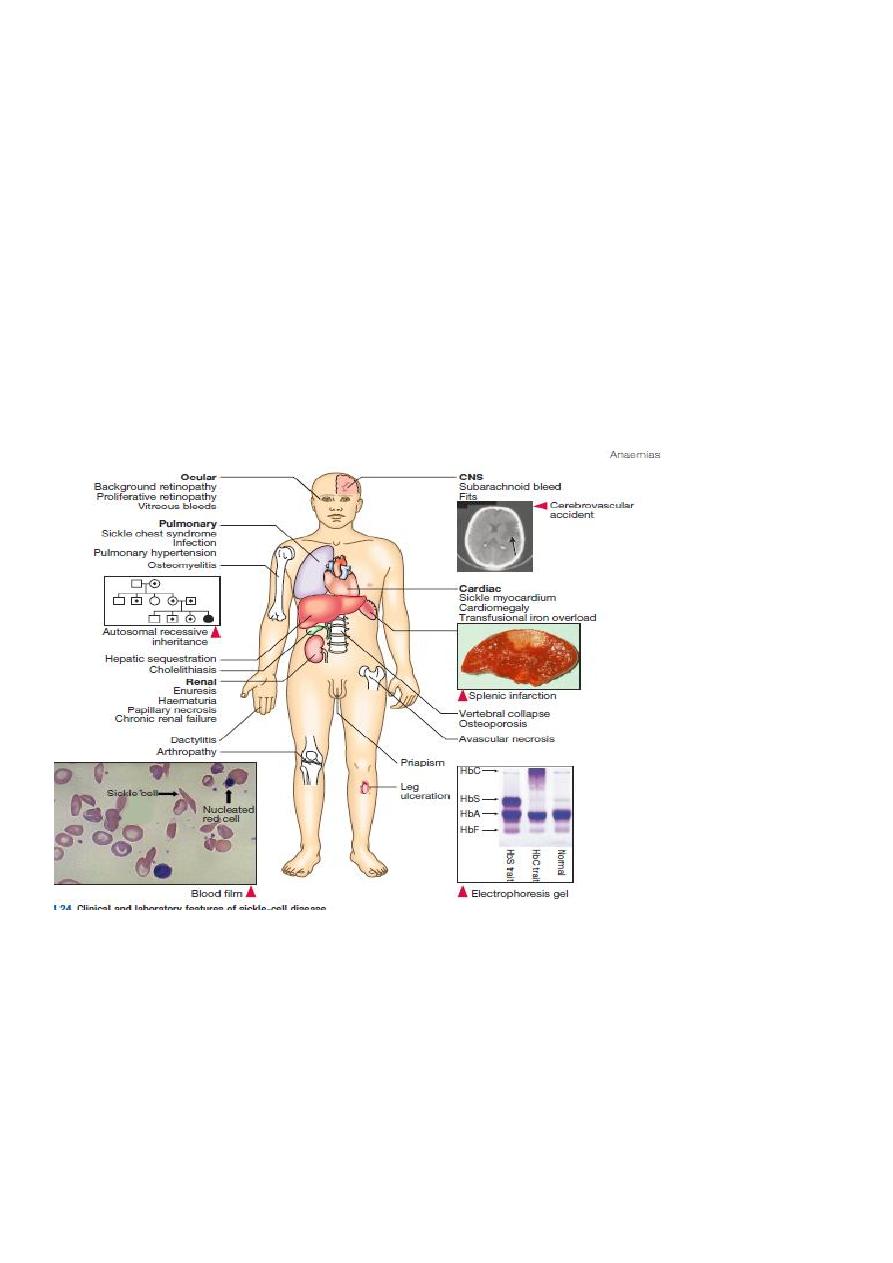
Sickle cell anemia:
It results from single base change in the DNA coding for the amino acid in the sixth
position in the b-globin chain.
This leads to an amino acid change from glutamic acid to valine
HbS will be formed
instead of the normal Hb.
Sickle Cell Anemia is a hereditary disease which is cause by a disorder in the blood, a
mutation in the Hemoglobin Beta Gene which can be found in the chromosome 11.
This disease causes the body to make abnormally shapes red blood cells. A normal red
blood cell
is shaped as a round donut while the abnormal red blood cell has a “ C “ form.
Hb S is insoluble and forms crystals when exposed to low oxygen tension.
Deoxygenated sickle Hb polymerizes into long fibrils.
The red cells sickle may block the different areas of the microcirculation or large
vessels causing infarcts of various organs.
It is widespread in Africa. Individuals with sickle-cell trait are relatively
resistant to the lethal effects of falciparum malaria in early childhood.
INTRODUCTION CONT’
Hemoglobin Beta Gene (HB-B) also known as Beta Globin is a protein that
resides in the red blood cells.
The HBB is 146 amino acids long and its molecular weight is 15,867
Daltons.
The molecules of the hemoglobin are responsible to carry oxygen
through the body.
2
The HBB is found in part 15.5 of the chromosome 11.
Pathophysiological effects of sickled cells
1.Extravascular hemolysis .
2. Loss of splenic function.
3.Anaemia.
4.Compromisation of the microcirculation.
CLINICAL MANIFESTATION
1.complication from moderate to severe anemia
2.slowed growth and development .
3.cardiac over load leads to CHF .
4.Bilirubin stones and cholecystitis .
5.Aplastic crisis.
Sickle cell crisis.
1.Splenic crisis (splenic sequestration syndrome, auto splenectomy)
2.Infections.
3.CNS and ophthalmic events (CVA, proliferative retinopathy).
4.Acute chest syndrome (chromic pulmonary hypertension lead to cor-pulmonale).
5.GIT : diffuse abdominal Pain.
6.Genitourinary symptoms:
- Painless haematuria.
- hyposthenuia.
3
- priapism.
- hypogonadism.
Sickle cell crisis Cont.
7.skeletal complication
- hand-foot syndrome.
- acute arthritis.
- aseptic necrosis of the head of femur.
- osteomyelitis .
8.Skin changes lead to chronic non-healing ulcer
DIAGNOSIS
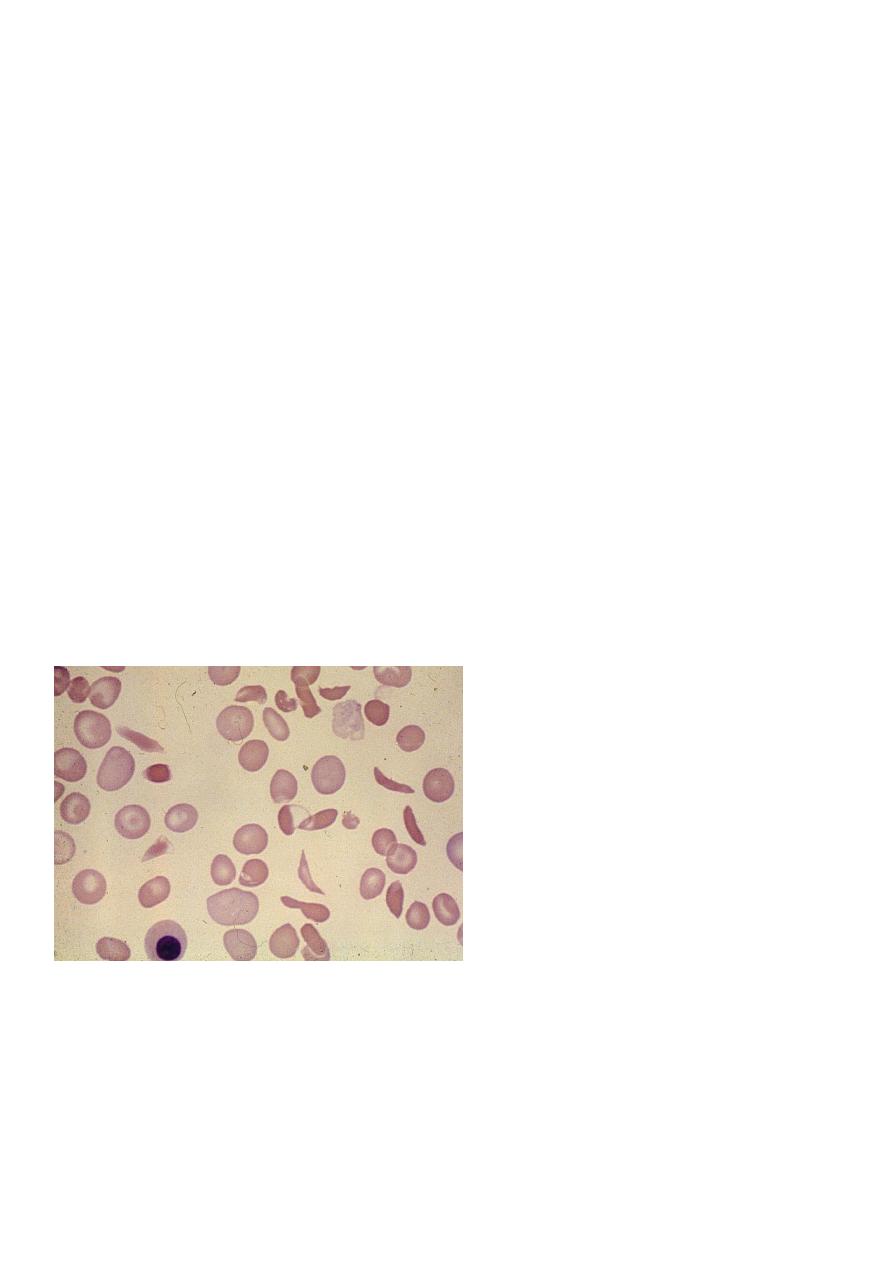
Peripheral blood smears :
sickled cells, target cells, Howell-Jolly bodies, normoblast, red cell fragment, increase
platelet and occasionally leukocytosis.
Screening test : sickling test
The presence of HbS can be demonstrated by exposing red cells to a reducing agent such as
sodium dithionite; HbA gives a clear solution,whereas HbS polymerises to produce a turbid
solution.
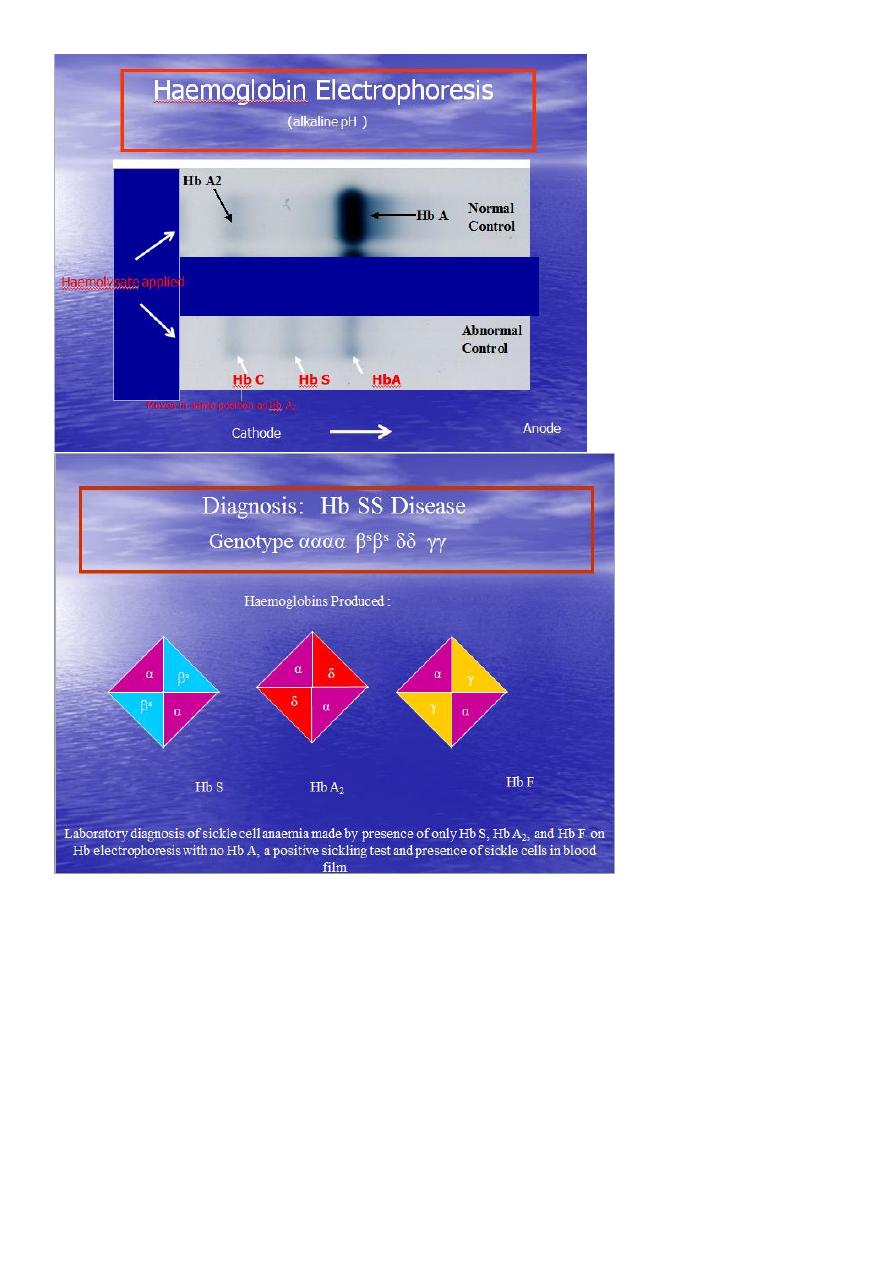
Definitive diagnosis by
Hemoglobin electrophoresis (Hb-S = 87% , Hb-F = 9.7%, Hb-A2 = 3.3%)
Howell Jolly Body Erythroblast
4
Diagnosis of sickle cell anaemia Cont.
Haemoglobin Electrophoresis:
Hb A 0 %
Hb S 87.0 %
Hb F 9.7 %
Hb A
2 3.3 %
Both parents of the affected individual will have sickle-cell trait

5
TREATMENT
Painful vaso-occlusive crisis
1.hydration
2.precepitating factors
3.oxygen therapy
4.analgesic
5.exchange transfusion
*Antisickling agent (Hydroxyurea) increase Hb F reduce sickling.
*Bone marrow transplantation(Allogeneic-BMT).
* Gene therapy.
Maintenance therapy and prevention
1.folic acid 1 mg/d orally.
2.pneumococcal vaccine.
3.pregnancy (increase crisis, abortion, stillbirth)
folate,, exchange transfusion. Exchange transfusion will increase Hb-A= 60%.
4.general anesthesia; Careful hydration and oxygenation.
*Angiographic contrast media causing sickling should be avoided
The thalassemia syndromes
Are inherited disorders arising from globin gene mutations that either reduce or totally
abolish the synthesis of one or more globin chains.
These imbalance in chain synthesis lead to formation of unstable Hb. or decrease Hb. lead
to hypochromic microcytic anaemia .
The thalassaemia named according to globin chain involved.
THALASSEMIA
Pathophysiology of thalassemia
α- thalassemia, gene deletions are responsible for the decrease or absence of α- chains.
ß- thalassemia : usually due to an mRNA abnormality.
This mutation reduces or eliminates the production of ß-globin chains
Clinical manifestations of thalassemia
ß- thalassemia major (ßoßo): is the most severe -
Becomes apparent 3-6 months after birth when switch from Hb-F to Hb-A takes place :
Hepatosplenomegaly (gall stones are also common )
Expansion of the bones (hair on end appearance on skull X-ray examination).
Severe anaemia with growth retardation and delayed sexual development

6
Damage to heart, pancreas, endocrines and liver due to iron over load
Pathogenesis of anaemia of ß-thala.
Decreased B- globin production has two major consequences :
total Hb. synthesis is reduced leading to
microcytic anaemia, low level of Hb-A lead to increase Hb-A2,Hb-F.
free α-chain accumulates in the RBC
α-chain accumulate and precipitate in RBC lead to hemolysis, destruction of RBC in the BM.
The extent to which ß-chain synthesis is suppressed determine the degree of anaemia
(ineffective erythropoiesis)
Extramedullary hematopoiesis and increased erythropoiesis in the BM lead to over all RBC
production is increased due to accumalation of α chain.
DIAGNOSIS OF ß-THALASSAEMIA
Positive family history with:
a.non specific findings :
Blood smear reveals microcytic RBCs, poikilocytosis, fragmented RBCs, MCV is low (around
65fl).
Heinz bodies are evident by supravital stains.
b. specific findings :
Definitive diagnosis of ß-thala. Is based on the following findings on Hb. Electrophoresis :
increased proportion of Hb-A2(> 3.5%).
increased proportion of Hb-F.

7
α –thalassaemia
Decrease synthesis of a-chains, lead to precipitation of Hb-H (4-ß chains) or Hb- Bart’s (4 δ-
chains)
Classification of α –thalassaemia:
1-Hydrops fetalis : severe, all 4 α -genes are deleted lead to severely anaemic, edematous,
stillborn infant. Hb-barts (4 δ -chains had very high oxygen affinity).
α-THALASSAEMIAS
2- HbH disease : deletion of 3 α –genes lead to unstable Hb. result in precipitation
and extra-vascular hemolysis.
3-α -thalassaemia Trait: deletion of 2 α genes.
4-α-thalassaemia Carrier: deletion of 1 α gene, asymptomatic.
DIAGNOSIS OF α-THALA
Positive family history with lab finding
non specific findings :
Blood smear show microcytic, hypochromic red cells, target cells, anisopoikilocytosis, and
decrease MCV.
Heinz bodies are evident.
Specific findings : definitive diagnosis is finding of HbH by Hb electrophoresis.

8
Hb –H Diagnosis
Haemoglobin Electrophoresis:
Hb A 91.5 %
Fast moving band 8.5%
Hb A
2
and Hb F decreased
Hb H Preparation
TREATMENT OF α- AND ß- THALASSEMIA
1.Regular red cell transfusions: hypertransfusion program (keep the level of Hb>110g/L)
2.Neocytes transfusion (increase RBC survival, decrease frequency of transfusion, and
decrease iron over load).
2-3 units every 4-6 weeks .
3.Leukocyte filter will lowers rate of transfusion reaction.
4.Folic acid supplementation (5 mg) to prevent aplastic crisis.
5.Iron chelation:
*Desferioxamine (Desferal) either with each unit of transfused blood (2 g) or by slow
subcutaneous daily infusions by pump (1-4g over 8-12)
*(Exjade ) Deferasirox
*Deferiprone
6.Splenectomy ;indication: mechanical difficulty,hypersplenism.
7.Bone Marrow Transplantation : prior to development of hepatomegaly, portal fibrosis &
iron over load.
PRENATAL DIAGNOSIS OF THALASSAEMIA
Guide parents and physicians in deciding
whether to complete pregnancy.
Both parent carriers.
Fetal diagnosis:
fetoscope to sample fetal venous blood show α/ß chain synthesis ratio.
Amniocentesis or trophoblast (chorionic villus) biopsy for DAN analysis using DNA probes.
IMMUNE HEMOLYSIS
Definition :
red cell life span is shortened because abnormalities in the components of the immune
system are specifically directed against the patients own erythrocytes.
1.Auto-immune hemolytic anaemia.
2.Transfusion related hemolysis.
3.Drug-related immune hemolysis.

9
AUTO- IMMUNE HEMOLYTIC ANAEMIA
The auto antibodies can be activated by either heat or cold.
Warm reactive auto immune hemolysis (37oC)
Causes :
1- idiopathic
2- secondary :
I. Drugs (Methyldopa)
II. Connective tissue disease (SLE)
III. Lymphoproliferative (CLL, HD, NHL)
CLINICAL MANIFESTATION
Onset rapid lead to anaemia, tiredness, fatigue.
Elderly pts. With atherosclerosis lead to chest pain.
Splenomegaly and Jaundice, may be absent in acute phase.
Abdominal pain and fever may also occur.
Diagnosis
spherocytosis, reticulocytosis, increase LDH, decrease serum haptoglolbin, increase indirect
bilirubin
positive direct coomb’s test; Patient’s CELLS are tested for surface Ab’s
Treatment
1.Removal of the underlying cause
2.Corticosteroid : 1mg/kg prednisone (3-4 weeks / check-Hb. & retics.) then slow tapering
if pt. respond . in chronic cases, use low dose therapy
3.Splenectomy : in case of steroid failure or decrease Hb. following cessation / reduction of
steroid.
4.Blood transfusion.
*cold-reactive auto immune hemolysis Auto Antibodies usually are IgM. Occasionally IgG.
Low temp make the antigen(Ag) more prominent on the membrane lead to antibodies
reaction.
Warm temp hiding the Ag below the membrane below the lipid component lead to
prevention of Ag-Antibodies(Ag-Ab.) reaction
CAUSES:
1.Idiopathic.
2.Secondary:
*Infection(mycoplasma pneumonia,
infectious mononucleosis)
*Lymphoma

10
Pathogenesis and clinical effects
The Blood from warm body cavities come to the surface low blood temp.this lead to
activation of the autoAbs and to agglutination resulting in impairing circulation
and producing cyanosis, pallor and ischemic pain (Raynauds phenomenon).
IgM binds Ag temporarily, but it fixes complement. IgM dis-engages from red cell to a
attach to another cell.
The fixed complement remains and activate C5-C6 causing hemolysis. C3-sensitized RBC
removed by kuppfer cells in the liver and this why hemolysis not respond to splenectom