
Abnormal development of
Female genital tractDr. Bara’a Lukman Humo Al-Ibrahim
FABOG/ FIBOG/ DGO
• Female genital abnormalities are uncommon and often do not present until, or well after, puberty.
• Anomalies due to Genetic Factors (Intersex)
• Anomalies Of The External Genital Organs (Ambiguous Genitalia)
• Anomalies Of The Internal Genital Tract "Müllerian Anomalies“
Anomalies of the internal genital tract "Müllerian anomalies"
Developmental anomalies of the müllerian duct system represent some of the most fascinating disorders that obstetricians and gynecologists encounter.A wide variety of malformations can occur when this system is disrupted. They range from uterine and vaginal agenesis to duplication of the uterus and vagina to minor uterine cavity abnormalities.
Müllerian malformations are frequently associated with abnormalities of the renal and axial skeletal systems, and they are often the first encountered when patients are initially examined for associated conditions.
Most müllerian duct anomalies are associated with functioning ovaries and age-appropriate external genitalia.
These abnormalities are often recognized after the onset of puberty. In the prepubertal period, normal external genitalia and age-appropriate developmental milestones often mask abnormalities of the internal reproductive organs. After the onset of puberty, young women often present to the gynecologist with menstrual disorders. Late presentations include infertility and obstetric complications.
Aetiology
The exact aetiology is unknown, the mostly accepted three theories are:
teratogenesis ,
genetic inheritance and
multifactorial expression.
Classification:
The most widely accepted method of categorizing müllerian duct anomalies is the AFS classification (1988). This system organizes müllerian anomalies according to the major uterine anatomic defect. It also allows for standardized reporting methods.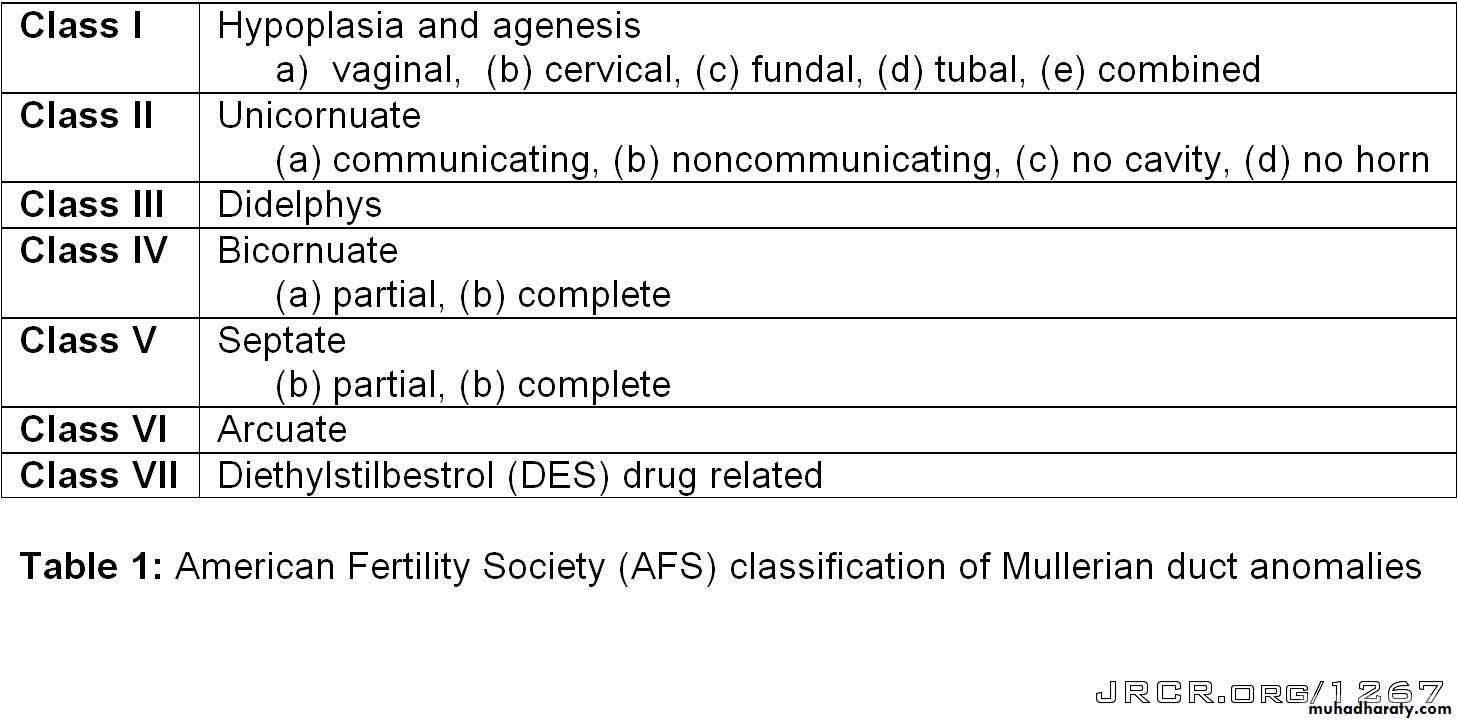
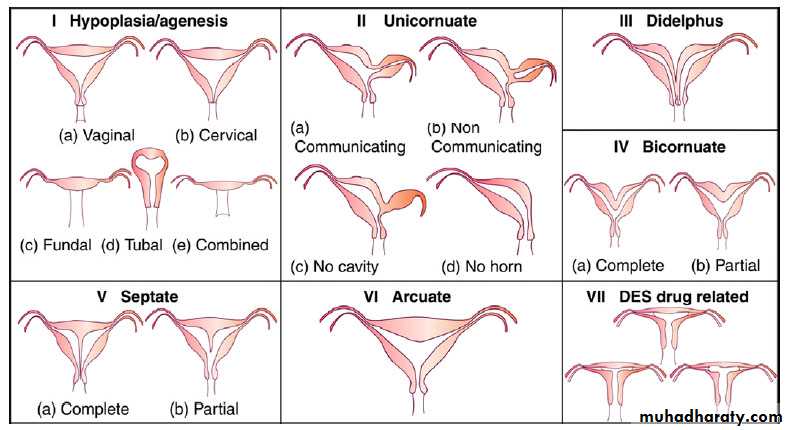
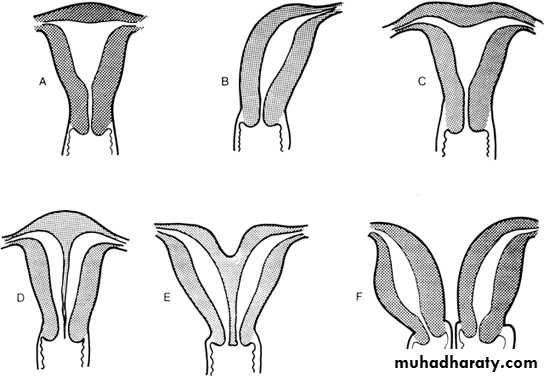
I: Segmental or complete agenesis or hypoplasia
Agenesis and hypoplasia may involve the vagina, cervix, fundus, tubes, or any combination of these structures. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome is the most common example in this category.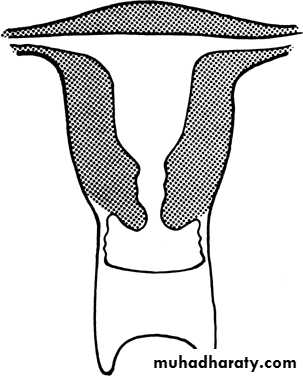
Vaginal atresia
a, Isolated congenital cervical atresia with normal vaginal development.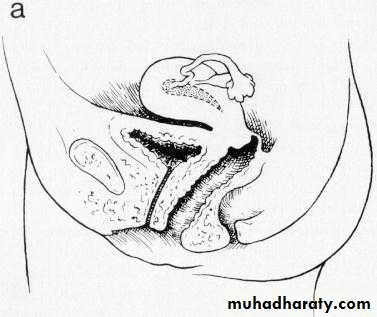
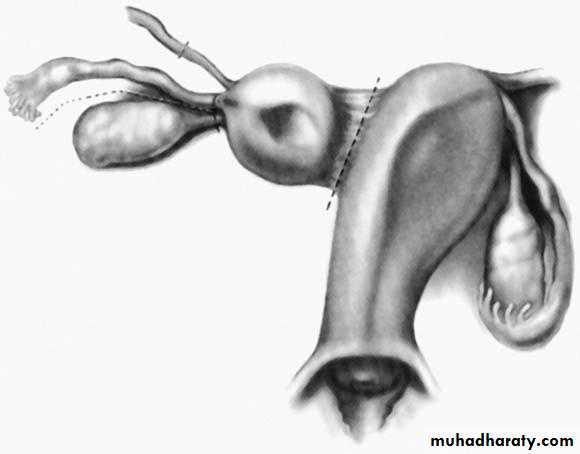
II:Unicornuate uterus with or without a rudimentary horn
When an associated horn is present, this class is subdivided into communicating (continuity with the main uterine cavity is evident) and noncommunicating (no continuity with the main uterine cavity). The noncommunicating type is further subdivided on the basis of whether an endometrial cavity is present in the rudimentary horn. The clinical significance of this classification is that they are invariably accompanied by ipsilateral renal and ureter agenesis.
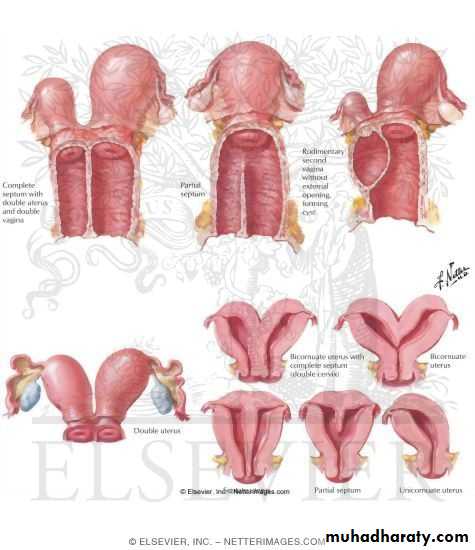
III: Didelphys uterus
Complete or partial duplication of the vagina, cervix, and uterus characterizes this anomaly.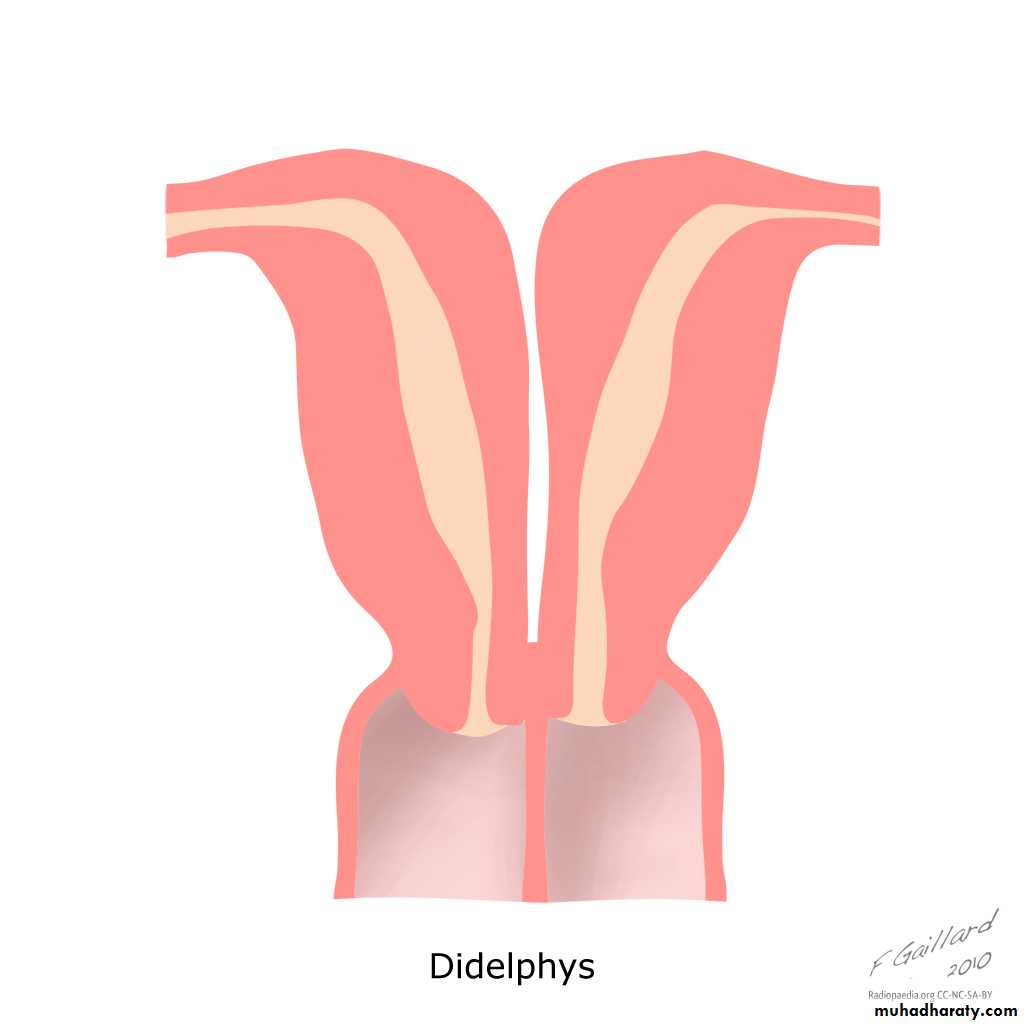
IV: Complete or partial bicornuate uterus
Complete bicornuate uterus is characterized by a uterine septum that extends from the fundus to the cervical os. The partial bicornuate uterus demonstrates a septum, which is located at the fundus. In both variants, the vagina and cervix each have a single chamber.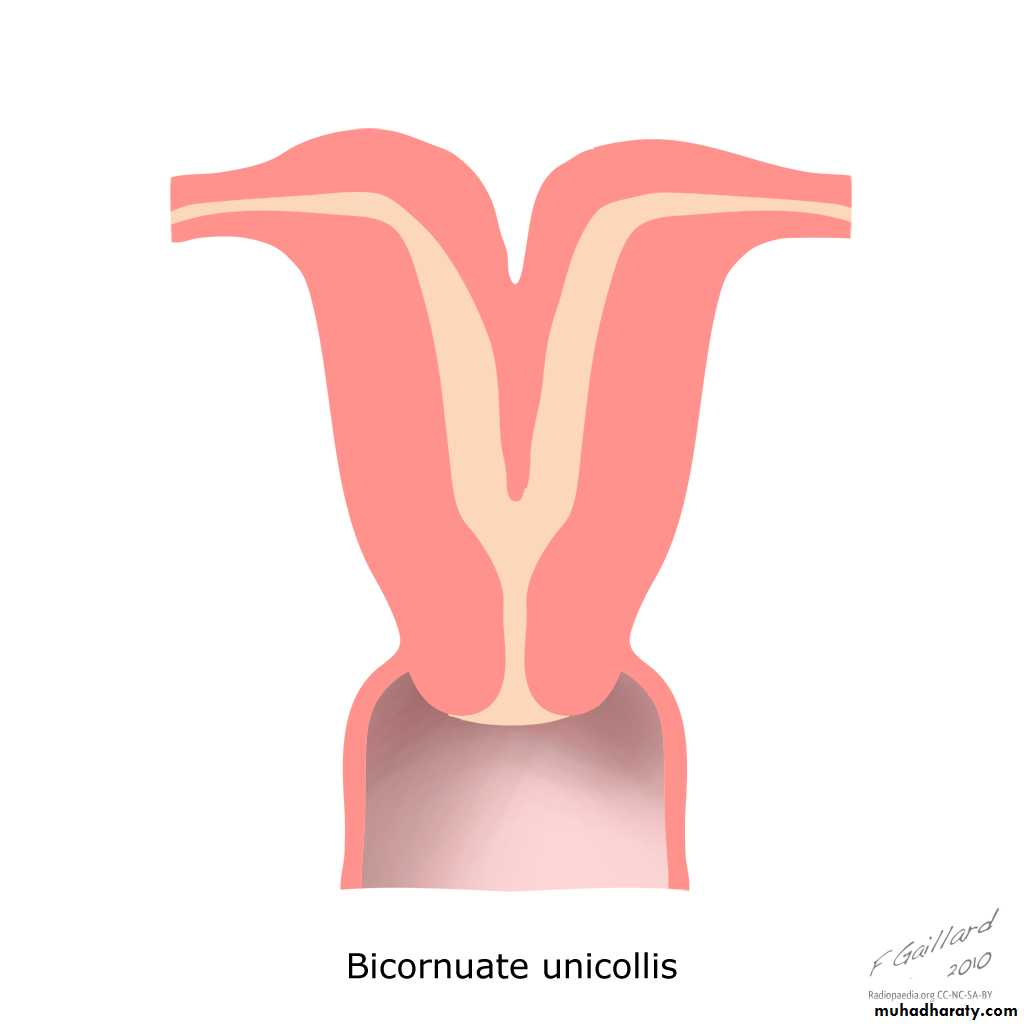
V: Complete or partial septate uterusA complete or partial midline septum is present within a single uterus.
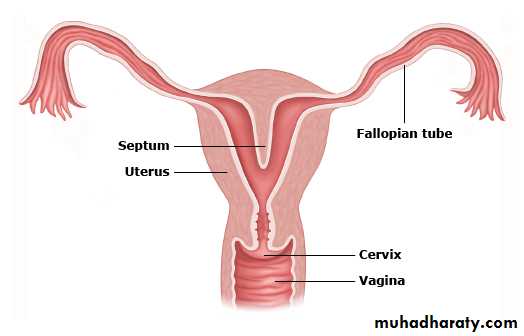
VI: Arcuate uterus
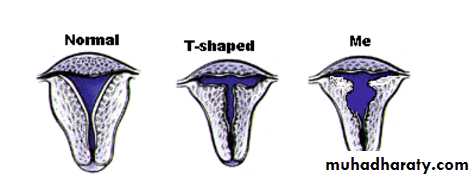
A small septate indentation is present at the fundus.VII: DES-related abnormalities
A T -shaped uterine cavity with or without dilated horns is evident.
Anomalies of the internal genital tract "Müllerian anomalies"
Obstructive Mullerian AnomaliesLongitudinal Fusion Anomalies
Agenesis/Hypoplasia And Other
Miscellaneous Anomalies
Presentation of uterine anomalies
-symptomless discovered during HSG or during operation.-dysmenorrhoea(rudimentary horn)
• -Complications during pregnancy and labour: late miscarriage, uterine rupture, premature labour, malpresentation, obstructed labour, retained placenta, postpartum haemorrhage.
-rare presentation is ectopic pregnancy in a rudimentary horn, presented later than other type and with rupture and sever haemorrhage.
Investigations:
Ultrasound.Hysterosalpingography, which allows evaluation of the uterine cavity and tubal patency.
MRI scan, which is considered the best imaging technique for uterine abnormalities.
Management:
Decision for surgical intervention will depend on the affect of the abnormality on enabling a viable pregnancy.
A septate vagina and the rudimentary horn of a bicornuate uterus are usually removed.
Uterine reconstruction is recommended for a bicornuate or septate uterus which is considered to be the cause of recurrent miscarriages.
Rudimentary horn attached to the unicornuate uterus with a band of tissue. Dashed lines represent the dissection planes.
Defects Not Classified by the AFSTransverse Vaginal Septum
TVS is formed when the tissue between the vaginal plate and the caudal aspect of the fused müllerian ducts fails to reabsorb. This anomaly divides the vagina into 2 segments, reducing its functional length.TVS can be perforate or imperforate, and can occur at nearly all levels in the vagina.
Most of these septa are located in the superior vagina (46%).
TVS is one of the most rare müllerian duct anomalies, with an approximate frequency of 1 case in 70,000 females.
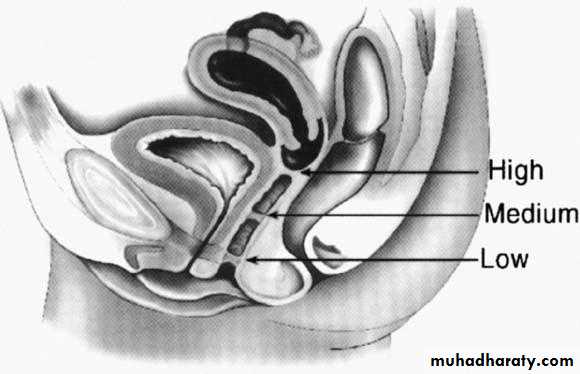
Potential sites of transverse vaginal septa. A. High septum. B. Midvaginal septum. C. Low septum.(From Simpson JL, Verp MS, Plouffe L Jr: Female genital system.
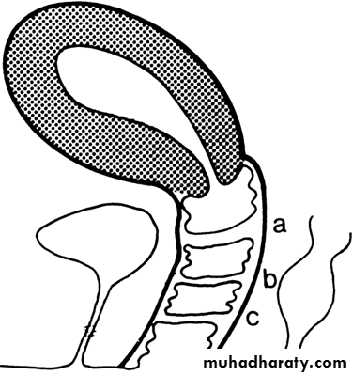
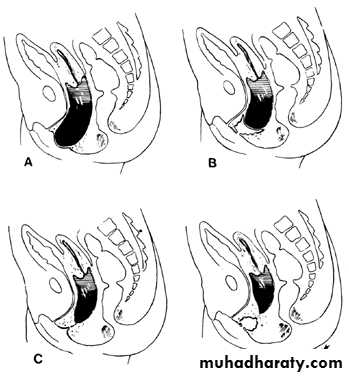
. Diagram of various lesions causing hydrometrocolpos.
A. Imperforate hymen. B. Transverse septum. C and D. Low and high atresia of vagina.Imperforate Hymen:
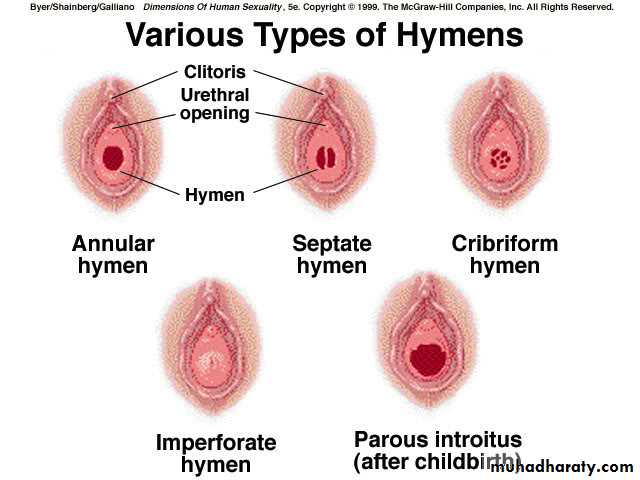
The hymen represents the junction of the sinovaginal bulbs with the urogenital sinus; hence it is formed form the endoderm of the urogenital sinus epithelium.The hymen is an area of tissue that represents the opening to the vagina. The hymenal tissue is a ring-like form of tissue which has a hole within the center, present at birth.
When no hymenal opening is present, a membrane covers the area of the hymen and this is called an imperforate hymen.
Imperforate hymen is likely the most frequent obstructive anomaly of the female genital tract, but estimates of its frequency vary from 1 case per 1000 population to 1 case per 10,000 population
Imperforate hymen is at the extreme of a spectrum of variations in hymenal configuration. Variations in the embryologic development of the hymen are common and result in fenestrations, septa, bands, microperforations, anterior displacement, and differences in rigidity and/or elasticity of the hymenal tissue.
Clinically:
May be discovered at birth ( "mucocolpos or hydrocolpos"More commonly at puberty: hematocolpos.
In the newborn period there may be a bulge of the hymenal membrane due to a blockage of the drainage of normal mucus from the baby’s vagina. The mother’s estrogen stimulates the production of mucus within the baby’s vagina and thus a white bulge may appear at the location of the normal opening to the vagina.
A young woman with an imperforate hymen which has not been surgically corrected will not have a normal menstrual period. This blockage may be associated with abdominal pain, back pain, or difficulty with urination.
Unfortunately, the typical findings at diagnosis may include a large collection of blood within the uterus (hematometra) and an even larger collection of blood within the distensible vagina (hematocolpos). Additional findings may include blood-filled fallopian tubes (hematosalpinges) and signs of retrograde menses, occasionally to the point of the development of intra-abdominal endometriosis and severe pelvic adhesions.
Treatment
Surgical repair after the onset of puberty but before menarche is optimal.This procedure should be performed using sterile technique in the operating room; after careful catheterization, a cruciate incision followed by excision of the four wedge-shaped tabs of hymen corrects the condition. Prophylactic antibiotics should be given postoperatively.
Medicolegal aspect in this condition is so important.
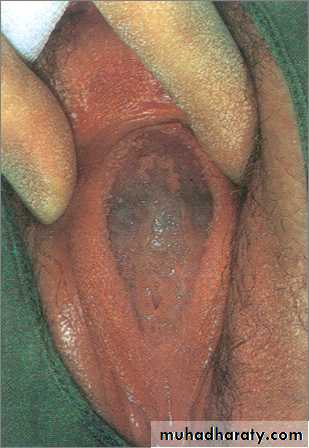
Imperforate hymen, classic appearance of bulging, blue-domed, translucent membrane.
Abdominal mass with imperforate hymen.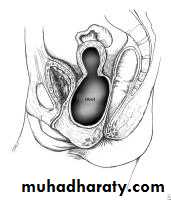
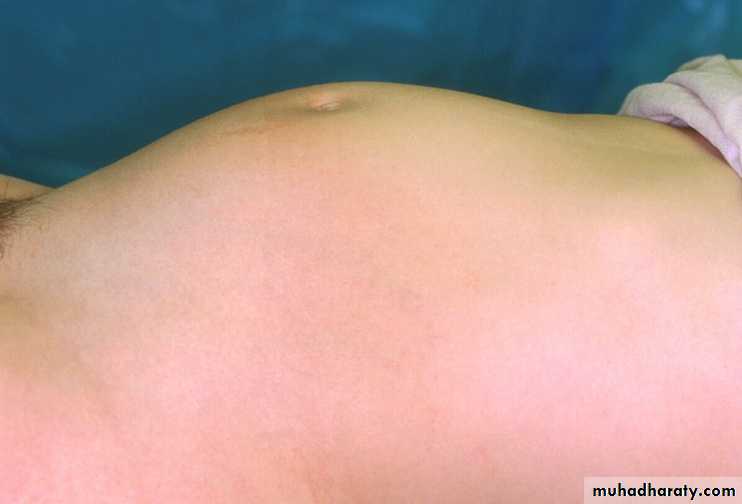
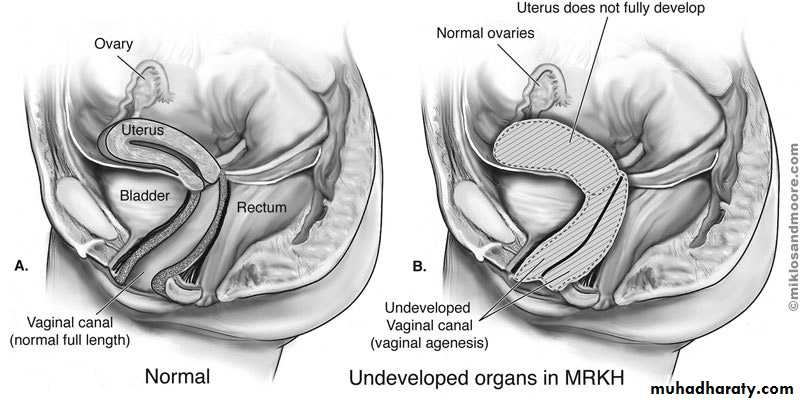
Mayer-Rokitansky-Kuster-Hauser Syndrome
The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is characterized by congenital aplasia of the uterus and the upper part (2/3) of the vagina in women showing normal development of secondary sexual characteristics and a normal 46, XX karyotype.MRKH may be isolated (type I) but it is more frequently associated with renal, vertebral, and, to a lesser extent, auditory and cardiac defects (MRKH type II or MURCS association(MÜllerian duct aplasia, Renal dysplasia and Cervical Somite anomalies) ).
Upper urinary tract malformations are found in about 40% of cases, including unilateral renal agenesis (23%-28%), ectopia of one or both kidneys (17%), renal hypoplasia (4%), and horseshoe kidney and hydronephrosis. Skeletal abnormalities are found in 10%, while auditory defects are found in 2% to 10% of cases.
Epidemiology
The incidence of MRKH syndrome has been estimated as 1 in 4500 female births.The majority of cases appears to be sporadic, however family cases have also been described. The mode of inheritance seems to be autosomal dominant with an incomplete degree of penetrance and variable expressivity, suggesting that the prevalence of the syndrome may probably be underestimated.
Type I (isolated) MRKH is less frequent than MURCS association
Clinical features:
The first sign of MRKH syndrome is a primary amenorrhea in young women presenting otherwise with normal development of secondary sexual characteristics and normal external genitalia, with normal and functional ovaries, and karyotype 46, XX without visible chromosomal anomaly. . At the same time, the vagina is reduced to a more or less deep (2–7 cm) vaginal dimple.Diagnostic methods:
Transabdominal ultrasonographyTransabdominal ultrasonograph is a simple and noninvasive method, and must be the first investigation in evaluating patients with suspected Müllerian aplasia. This technique reveals an absence of the uterine structure between the bladder and the rectum.
Magnetic resonance imaging (MRI)
MRI is a non-invasive technique that provides a more sensitive and more specific means of diagnosis than ultrasonography. It should be performed when ultrasonographic findings are inconclusive or incomplete. MRI allows an accurate evaluation of the uterine aplasia, as well as a clear visualization of the rudimentary horns and ovaries. Moreover, MRI can be used at the same time to search for associated renal and skeletal malformations.
laparoscopy
This is an invasive technique requiring hospitalization and anesthesia. It is performed in cases of doubtful diagnosis after ultrasonography and/or MRI. Laparoscopy is not only useful for diagnosis of uterine malformations but also for assisted vaginoplasty.Once MRKH syndrome is diagnosed, a full check-up must be undertaken to search for associated malformations. Since renal and skeletal abnormalities may not be symptomatic, it is necessary to perform at least transabdominal ultrasonography and spine radiography. In case of suspicion of hearing impairment and/or a cardiac anomaly, complementary audiogram and/or heart echography must also be carried out.
Differential diagnosis:
Differential diagnosis of Müllerian aplasia includes patients presenting with primary amenorrhea and with normal secondary sexual characteristics.This should first lead to exclusion of gonadal dysgenesis.
The differential diagnosis includes congenital absence of uterus and vagina (aplasia or agenesis), isolated vaginal atresia and androgen insensitivity.
Transverse vaginal septum and imperforate hymen, which can be initially misleading, are not included. Indeed, patients with these latter conditions have normal cervix and uterus, both of which are palpable on rectal examination. Ultrasonography can be used to define the Müllerian structures in infrequent cases where palpation is unrevealing.
Management
Young women diagnosed with MRKH syndrome suffer from extreme anxiety and very high psychological distress when they are told they have no uterus and vagina. Thus, it is recommended that the patient and family attend counseling before and throughout treatment.Treatment consisting of creating a neovagina must be offered to patients only when they are ready to start sexual activity and also when they are emotionally mature.
Treatment may be either surgical or nonsurgical, but the chosen method needs to be tailored to the individual needs, motivation of the patient and the options available.
Nonsurgical creation of a neovagina
The most commonly used nonsurgical procedure is Franck's dilator method. It involves the application, first by the clinician and then by the patient herself, of vaginal dilators (Hegar candles), progressively increasing in length and diameter.Dilators are placed on the perineal dimple for at least 20 minutes a day. The whole process takes between six weeks and several months, with a success rate varying from 78% to 92%.
As this nonoperative approach is noninvasive and often successful, it is recommended as a first-line therapy. However, it can be applied only when the vaginal dimple is deep enough (2–4 cm).
Surgical creation of a neovagina
McIndoe operation: this involves the dissection of a space between the rectum and the bladder, placement of a mold covered with a skin graft into the space, and diligent postoperative vaginal dilatation.
Ultimately, infertility will be the most difficult aspect of the disorder for the patient to accept. Assisted fertility techniques, including surrogacy, enable women without a uterus to have genetic offspring.