Hemorrhagic Disorders..
These includeDisorders of platelets.
Disorders of blood vessels.
Disorders of coagulation & fibrinolysis.
Platelet Disorders
Quantitative : Thrombocytopenia.Qualitative : Platelet defects (functional anomalies).
Thrombocytopenia
Thrombocytopenia exists when platelet count is less than 150 x 109 /L .Normal platelet count = 150 – 400 x 109 /L
Bleeding is unusual when count is >50x109 /L
Spontaneous bleeding occurs when count is < 20x109 /L
Causes of Thrombocytopenia
1.decresed platelet production
Characterized by reduction of megakaryocytes in bone marrow & by small mean size of circulating platelets (Mean Platelet Volume –MPV ) and association with anemia and leucopenia :Aplastic anaemia.
Megaloblastic anaemia ( decrease Vit. B12 or /and decrease folic acid ).
Bone marrow infiltration by neoplasms.
Cytotoxic drugs ( Dose Dependant ).
Ionizing radiation (Dose Dependant ).
Drugs; cause thrombocytopenia in some recipients :Metheprim, Phenylbutazone, Gold compounds .
Alcohol.
2. Increased destruction of platelets
Characterized by normal or increased numbers of megakaryocytes in bone marrow , circulating platelets appear larger than normal ( raised MPV) and that platelets are usually only affected ( no anaemia or leucopenia ).
Causes of Increased Destruction of Platelets :
hypersensitivity to drugsOccurs suddenly following single dose drugs act as a hapten forming antigenic complex by binding to plasma protein and then antibody ( usually IgG) is formed against this complex , this antigen-antibody complex then binds to platelets leading to destruction by phagocytosis usually in the spleen .
Drugs : Chlorothiazides , Digoxin , Methyl- dopa ,PAS ( para-aminosalicylic acid ), Quinine, Quinidine, Sulphonamides .
Autoimmune Thrombocytopenia
Autoantibodies usually of IgG class either asisolated disorder :idiopathic (immune ) thrombocytopenic purpura ( ITP)
in association with other autoimmune disorders : SLE ,myasthenia gravis ,Evan’s syndrome( autoimmune hemolytic anemia + autoimmune thrombocytopenia), lymphoma , chronic lymphocytic leukaemia .

ITP (Idiopathic ((Immune)) Thromocytopenic Purpura)
Occurs chiefly in children and young adultsResponsible antibody usually belongs to subclass 3 of IgG.
ClinicallyVaries from mild cutaneous bleeding to gross uterine or GIT hemorrhage .
In severe cases it lead to intracerebral hemorrhage .
Treatment
SteroidsImmunosuppressive drugs
Splenectomy
3. Hypersplenism
Clinical syndrome :Enlargement o f the spleen.
Reduction in one or more of cell lines of blood (anemia, leucopenia, thrombocytopenia).
Normal bone marrow.
Cure after splenectomy.
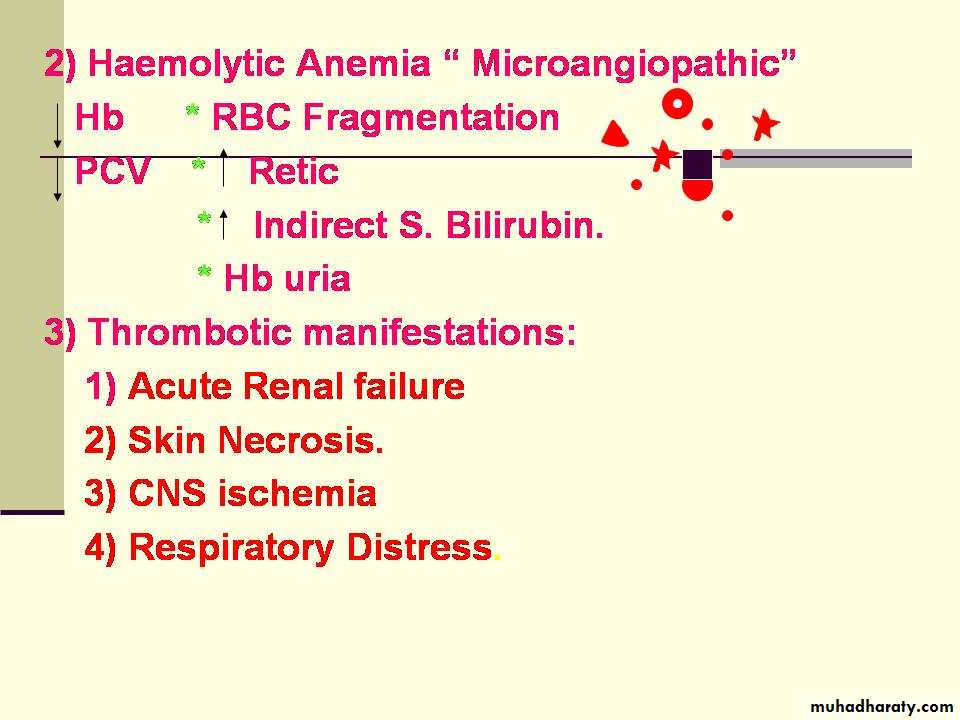
4.DIC(disseminated intravascular coagulation)
This causes thrombocytopenia by excessive utilization & destruction of platelets .
5. Massive blood transfusion
Qualitative Platelet Defects
Platelet count is normal ,but there is defect in platelet aggregation .e.g. Glanzmann’s disease (thrombosthenia, autosomal recessive)
Disorders of Blood Vessels ( Vascular Purpra )
Congenital :Hereditary Hemorrhagic Telagiectasia
Autosomal dominant
Clinically: usually epistaxis , multiple telangiectatic spots in the skin & mucus membranes leading to hemorrhage & iron deficiency anemia ,haemoptysis.
Acquired :
1)Purpura simplex in women .
2)Senile purpura :on the dorsum of hands & arms due to poor capillary support from collagen as also in :
3)Steroid therapy or Cushing syndrome
4)Scurvy ,vit. C needed for polymerization of mucopolysaccharides necessary for collagen synthesis .Henoch Schonlein Purpura : necrotizing vasculitis give rise to small hemorrhages especially in the skin & gut ,there may be associated glomerulonephritis ,usually follow streptococcal infection.
Damage to capillaries as in :
severe acute bacterial infection: septicaemia.
subacute bacterial endocarditis .

Inherited Disorders of Coagulation
Of these coagulation factors deficiencies factor VIII deficiency is important .it can lead to Haemophilia A and von Willebrand’s disease .Structure of factor VIII
Plasma factor VIII is now considered to be a complex of two components ;the larger of the two ,factor VIII /von Willebrand factor ( VIII R: WF) is coded by autosomal genes and is deficient in von Willebrand ‘s disease , it promotes primary haemostasis by interacting with platelets and also appears to function as a carrier of smaller component factor VIII coagulant (VIII C) which is coded by an X chromosome which participates directly into cascade clotting reaction & is deficient in classical haemophilia ,when assayed immunologicaly these two components are expressed as antigen (Ag)i.e. VIII R: Ag and VIII C : Ag .
Haemophilia A
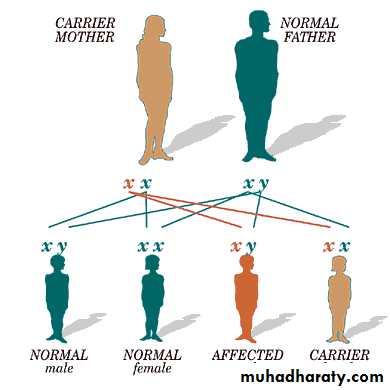
Hereditary abnormality of coagulation.Sex linked : affect ♂ ,while ♀ are carriers .
Xْ Y XX
YX YX Xْ X Xْ X
Normal ♂ Carrier ♀50% of daughters of carrier female are carriers .
50% of sons of carrier female are diseased .XY Xْ X
XXْ XX YXْ YXHow do you get it ctd.
Clinically
Male child will suffer from bleeding following circumcision , haemarthrosis usually after crawling .Severity of haemophilia is graded according to the level of VIII C into:
Severe ( VIII C < 1% of normal ).
Moderate ( 2-5% of normal).
Mild ( 5-20% of normal).
This is a diagram of the joints most commonly affected by Hemophilia. It most often occurs at the knees, hips, ankles, shoulders, and elbows

Diagnosis
APTT ↑Bleeding time normal
VIII C activity ↓
VIII C : Ag ↓
VIII R: Ag normal
Von Willebrand’s Disease
Inherited hemorrhagic disease in which bleeding time is prolonged due to deficiency of von Willebrand’s factor (vllll R) as this factor is important for platelet adhesion to vascular subendothelium.
Factor IX deficiency ( Haemophilia B or Christmas Disease )
Inherited disorder shows the same pattern of inheritance as haemophilia A (sex linked ).Same clinical picture but incidence of disease = 1/5th of the haemophilia A .
Treated by factor IX concentrate .
Acquired Disorders Of Coagulation
Vitamin K deficiency
Vitamin K is necessary for γ carboxylation of precursors of factor II ( prothrombin ) & some other coagulation factors. It is fat soluble ,present in leaf vegetables & also synthesized by the normal intestinal flora.
Dietary deficiency of sufficient severity to produce bleeding is well recognized in:
Neonates (Haemorrhagic Diseases of the newborn) in whom normal bacterial flora is not yet established.
In children & adults( malnourishment).
↓ absorption in billiary obstruction, coeliac disease.
Liver disease
Liver is the site of synthesis of most coagulation factors.Severe impairment of liver lead to combined factor deficiency particularly II , VII ,IX ,X, &
I (fibrinogen).
Renal Impairment
Lead to thrombocytopenia ,platelet dysfunction ,(II ,VII ,IX ,X ,XIII ) ,DIC (haemolytic uraemic syndrome).Warfarin therapy
Oral anticoagulant act as competitive inhibitor of vit. K ,suppressing the synthesis of four vit. K dependant clotting in the liver prothrombin ( factor ll ,VII ,IX & X .)Control of Warfarin Therapy by:
Doing prothrombin time
Control = seconds.Test = seconds.
Test/control ratio (R) =
INR (international normalized ratio ) =
Accepted INR = 2 - 3.5
INR = (R)^s
S= sensitivity index ,fixed figure provided by manufacturer of the kit ( e.g S = 2)
Heparin therapy control
Coagulation ( Clotting ) timeThrombin time
Activated Partial Thromboplastin Time (APTT)
Disseminated Intravascular Coagulation (DIC)
wide spread deposition of fibrin in the small vessels of many organs causing tissue necrosis & multiple organ dysfunction and subsequent bleeding state due to consumption of platelets & clotting factors and secondary enhancement of fibrinolytic activity . Microangiopathic haemlytic anaemia is a common accompaniment.Causes of DIC
Extensive burnSepticaemia
Shock
Liver disease
Renal disease
Complications of labour : retroplacental haemorrhage & aminotic fluid embolism.
Malignancies , leukaemia especially acute promyelocytic leukaemia (M3 in FAB classif.)
DIC: Disseminated Intravascular Coagulation: